
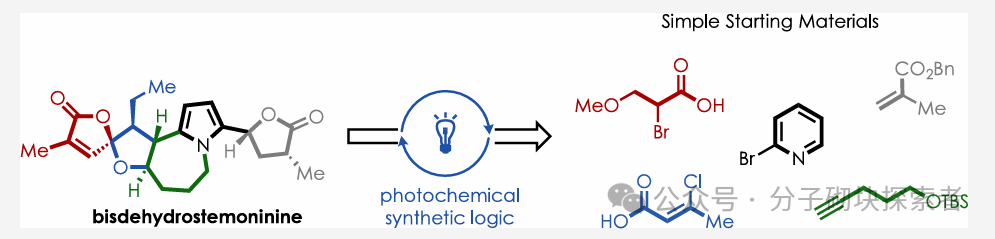
 Image source: J. Am. Chem. Soc.Introduction:
Image source: J. Am. Chem. Soc.Introduction:
Recently, Professor David A. Nicewicz and his research group at the University of North Carolina at Chapel Hill reported a novel synthetic strategy utilizing iterative photochemical methods, providing mild reaction conditions and a straightforward synthetic procedure to achieve the total synthesis of three structurally complex stemoamide alkaloids.
The key retrosynthetic analysis involved the dual oxidation and reduction capabilities of an acridinium photoredox catalyst, constructing a densely functionalized tetrahydrofuran ring through a polar radical cross-cyclization reaction (PRCC). The resulting lactone was then used as a functional handle for further polar radical cross-cyclization reactions to construct a unique oxazabicyclobutene structure. Finally, a late-stage heteroaromatic transformation yielded a key intermediate for the synthesis of the aforementioned three stemoamide alkaloids.
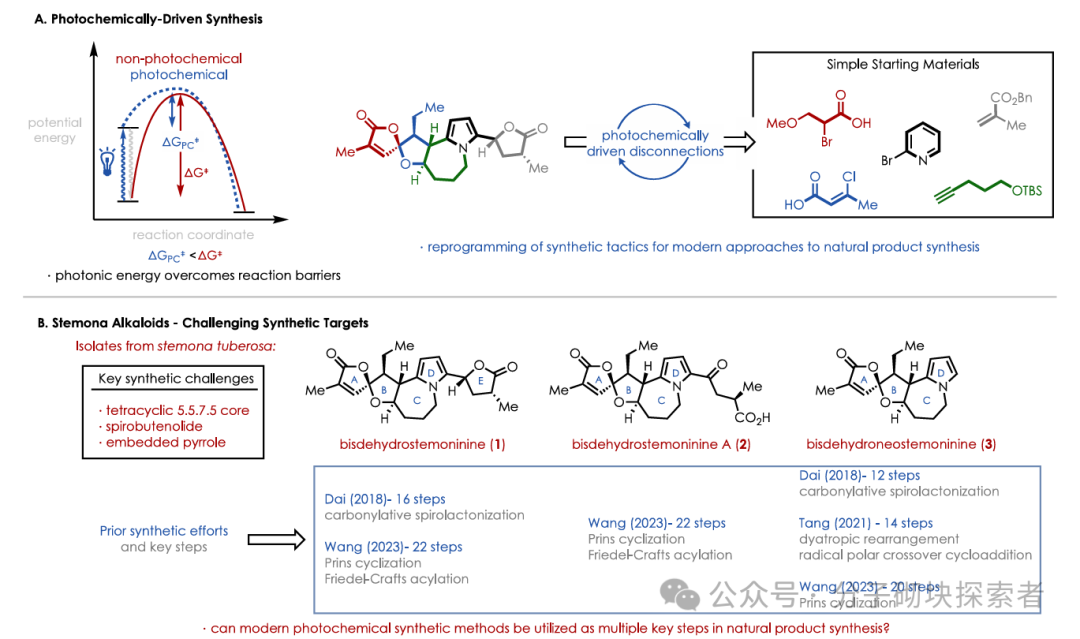 Image source: J. Am. Chem. Soc.
Image source: J. Am. Chem. Soc.
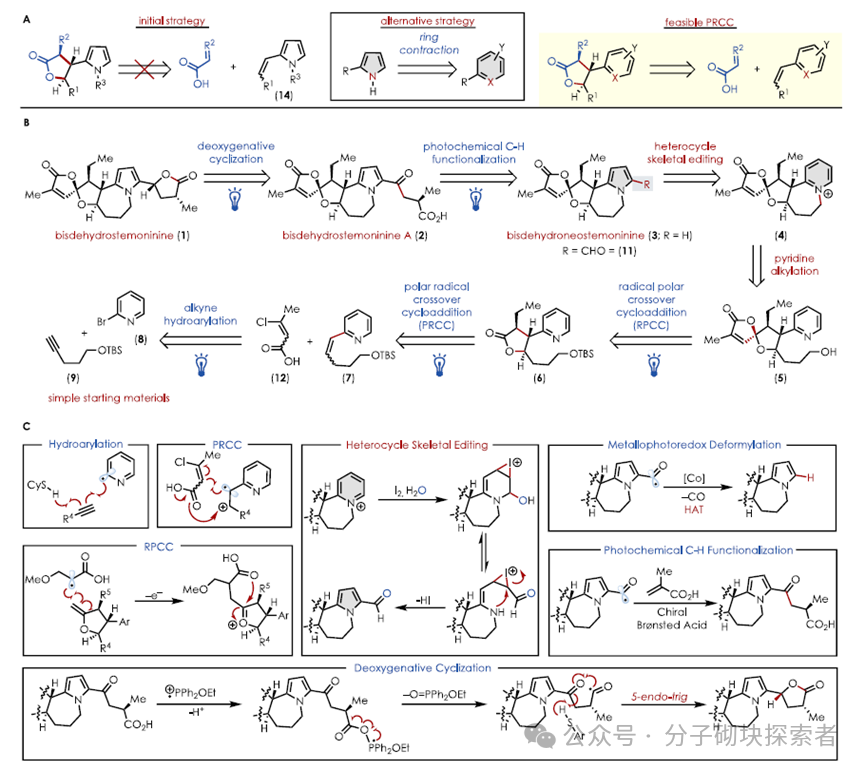
Due to the incompatibility of β-substituted vinyl pyrrole 14 with the critical PRCC reaction conditions, the authors previously demonstrated that vinyl (hetero)arenes are compatible in the PRCC reaction system. Thus, by ring-opening/closing contraction, the pyridinium was converted to 2-formylpyrrole using literature methods, selecting the reactive compatible 2-vinylpyridine as a synthetic equivalent for pyrrole. The photochemical PRCC reaction successfully constructed the core skeleton, followed by an efficient late-stage pyridinium salt to 2-formylpyrrole ring contraction rearrangement, transforming the pyridine ring into the target pyrrole ring, masking the pyrrole as pyridine to overcome the limitations faced by the oxidative photochemical reactivity of pyrrole.
The vinyl pyridine 7 was obtained through a photoredox-catalyzed hydrogenation reaction reported by the Jui group. When subjected to a PRCC reaction with crotonic acid, a reverse Markovnikov hydromethoxylation reaction occurred, failing to yield the expected lactone. This may be due to the polarization of the crotonic acid ester olefin, requiring a β- substituent to polarize the olefin towards the desired cyclization mode, leading to the introduction of β-chloro crotonic acid 12. The desired lactone 6 was obtained through subsequent elimination-reduction sequences.
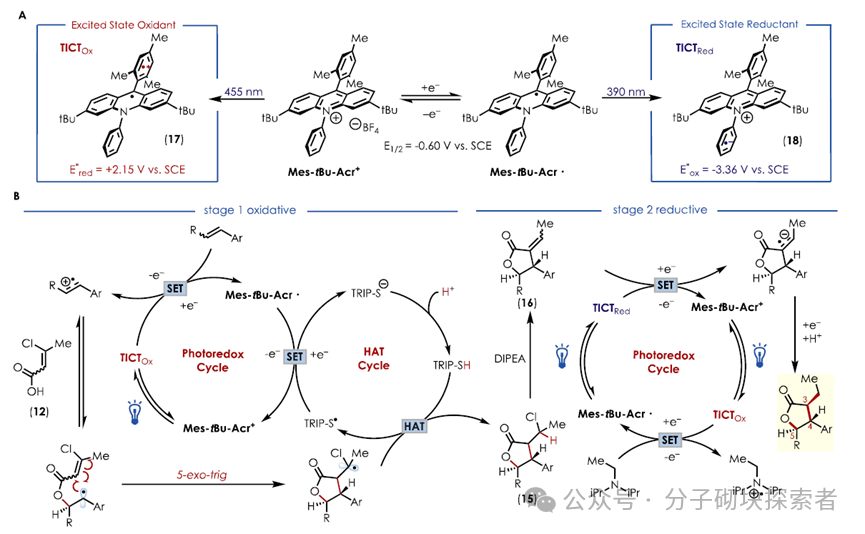
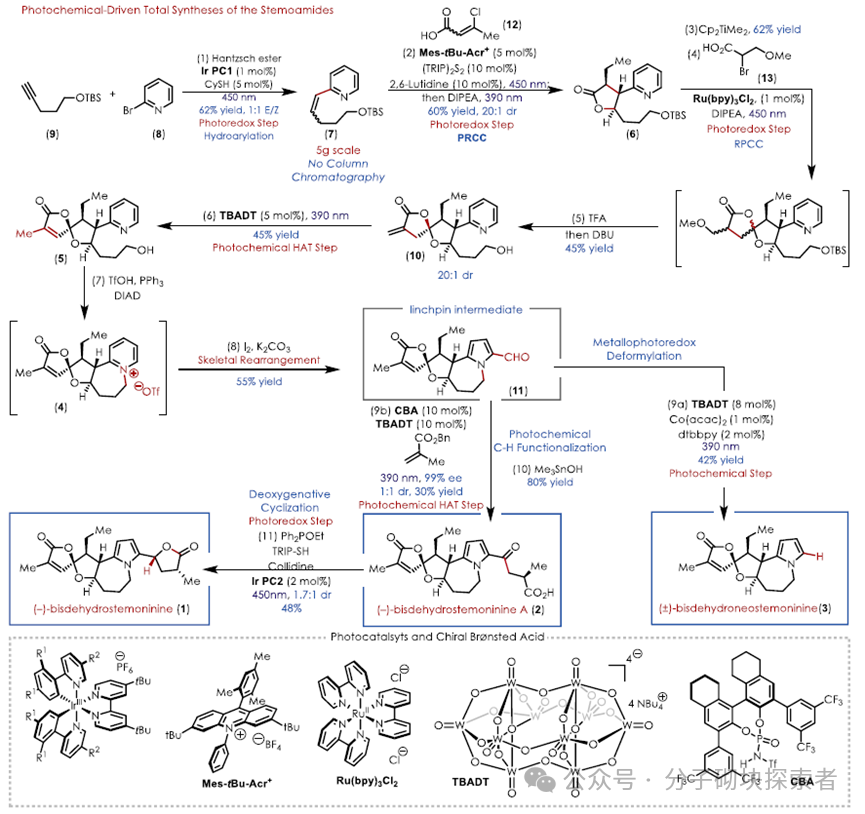 Utilizing the dual functionality of the photocatayst Mes-tBu-Acr⁺ , a one-pot method was employed, where the olefin 7 was single-electron oxidized to a cationic radical under the catalyst’s oxidized state, and in synergy with the HAT catalyst, reacted with acid 12 to undergo a PRCC reaction, yielding the chlorinated intermediate 15. Upon adding base DIPEA and changing the light wavelength (390 nm), DIPEA facilitated the elimination reaction, generating 16 to obtain the α,β-unsaturated lactone 16. The DIPEA acted as an electron donor, reducing the catalyst to its reduced state (TICTᵣₑ𝒹), thereby reducing the olefin in 16 and ultimately yielding the lactone 6. This reaction exhibited complete regioselectivity and excellent enantioselectivity, likely stemming from the intrinsic stereocontrol of the PRCC and the excess base promoting subsequent diastereomerization, successfully constructing the three consecutive chiral centers required for the B ring.
Utilizing the dual functionality of the photocatayst Mes-tBu-Acr⁺ , a one-pot method was employed, where the olefin 7 was single-electron oxidized to a cationic radical under the catalyst’s oxidized state, and in synergy with the HAT catalyst, reacted with acid 12 to undergo a PRCC reaction, yielding the chlorinated intermediate 15. Upon adding base DIPEA and changing the light wavelength (390 nm), DIPEA facilitated the elimination reaction, generating 16 to obtain the α,β-unsaturated lactone 16. The DIPEA acted as an electron donor, reducing the catalyst to its reduced state (TICTᵣₑ𝒹), thereby reducing the olefin in 16 and ultimately yielding the lactone 6. This reaction exhibited complete regioselectivity and excellent enantioselectivity, likely stemming from the intrinsic stereocontrol of the PRCC and the excess base promoting subsequent diastereomerization, successfully constructing the three consecutive chiral centers required for the B ring.
The lactone 6 was reacted with Petasis reagent via an olefination reaction to yield an exo-vinyl ether, which underwent a polar radical cross-cyclization reaction with α-bromo carboxylic acid 13 under photochemical conditions, initially generating a mixture of four diastereomers. Subsequently, a tandem acid/base treatment efficiently transformed the complex mixture into a single isomer, yielding intermediate 10.
Intermediate 10 required harsh conditions and rare metal catalysts for traditional isomerization of the exo-olefin, while the authors innovatively employed tetrabutylammonium decatungstate (TBADT) as a hydrogen atom transfer (HAT) reagent, successfully achieving this transformation under visible light, overcoming the challenges posed by multiple weak C-H bond competitive reactions in intermediate 10, and successfully obtaining the isomerization product 5.
Utilizing a mild Mitsunobu reaction to replace traditional intramolecular nucleophilic substitution, the authors efficiently constructed the pyridinium salt ring system. The extraction and separation allowed for a direct one-pot reaction of the pyridinium salt to form 2-formylpyrrole, yielding the key intermediate 11.
To complete the total synthesis of bisdehydroneostemoninine (3), a deformylation reaction of 11 was required. Traditional methods necessitated the use of superstoichiometric Wilkinson catalyst ([RhCl(PPh₃)₃]) and high temperatures, which were harsh conditions. Inspired by the synergistic de-formylation reaction of a light-activated HAT catalyst with a cobalt catalyst, the authors explored a photochemical alternative. Initial attempts using literature conditions were unsuccessful, but they later discovered that tetrabutylammonium decatungstate (TBADT) as a HAT catalyst could efficiently promote this reaction, achieving the shortest synthesis sequence for bisdehydroneostemoninine (3 ) to date.
Bisdehydrostemoninine A (2) needs to be generated from the key intermediate 11 through selective formyl C-H functionalization, while the formylpyrrole structure is incompatible with conventional Stetter reactions using N-heterocyclic carbene (NHC) catalysts. Inspired by the cooperative asymmetric Stetter reaction using chiral Brønsted acids (CBA) and TBADT, the authors screened and found that chiral phosphoramidite catalysts (CBA C), with a pKₐ of 6, could optimally cooperate with TBADT. Implementing this stereodivergent transformation on (±)-11 yielded a non-racemic mixture of 1:1 (ee value reaching 99%), and through kinetic resolution, a 15% yield of the desired non-racemic product was obtained from a maximum theoretical yield of 50%. Subsequently, mild hydrolysis of the ester group using Me₃SnOH (to avoid the diastereomerization caused by strong bases) yielded bisdehydrostemoninine A (2).
To achieve the final synthetic target bisdehydrostemoninine (1), the authors employed the deoxygenative 5-endo-trig cyclization reaction reported by Doyle et al. to complete the crucial final step of reductive lactonization. This approach exhibited chemical selectivity, replacing traditional polar reducing agents, ultimately achieving the total synthesis of bisdehydrostemoninine (1) in 11 steps, creating the shortest chemical synthesis route for this molecule to date.
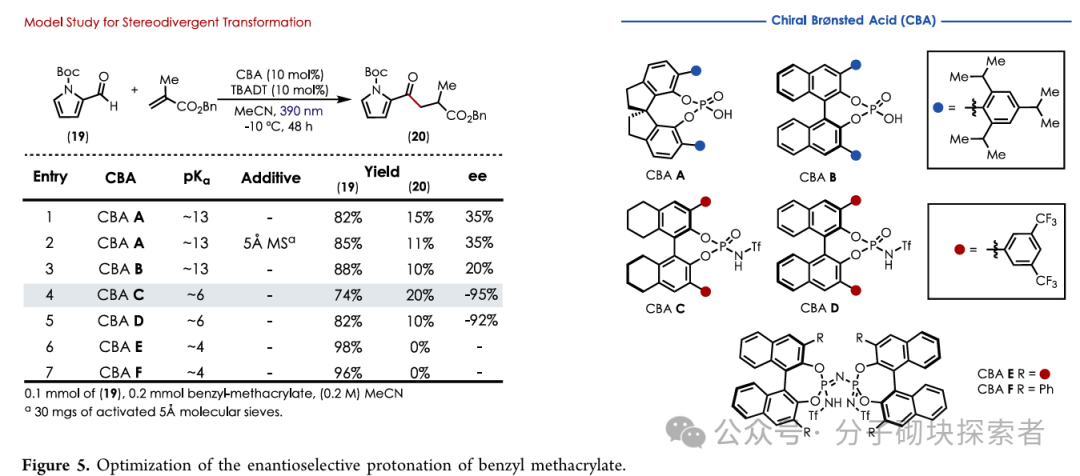 Image source: J. Am. Chem. Soc.
Image source: J. Am. Chem. Soc.
Conclusion:
This article reports on the iterative photochemical total synthesis of three stemoamide alkaloids, centered around photochemical and photoredox steps. This strategy relies on the latest reactivity in photochemical/photoredox methods, providing a unique synthetic approach. Utilizing the dual oxidation and reduction capabilities of the Mes-tBu-Acr⁺ photocatalyst, a densely functionalized bicyclobutene-lactone-tetrahydrofuran A-B ring system was successfully constructed in a five-step sequence, common to stemoamide alkaloids. By employing a photochemical retrosynthetic analysis strategy and leveraging the characteristics of highly reactive species, the shortest synthesis route for this class of stemoamide alkaloids was developed to date. The entire sequence avoided traditional redox operations and significantly minimized the use of protecting groups. Furthermore, this synthetic scheme opens up possibilities for obtaining other stemoamide alkaloids containing pyrrolidine rings instead of pyrrole rings through subsequent additional redox modifications.
References:
Photochemically Enabled Total Syntheses of Stemoamide Alkaloids
J. Am. Chem. Soc.2025, 147, 18, 15482–15489
https://pubs.acs.org/doi/10.1021/jacs.5c01788.