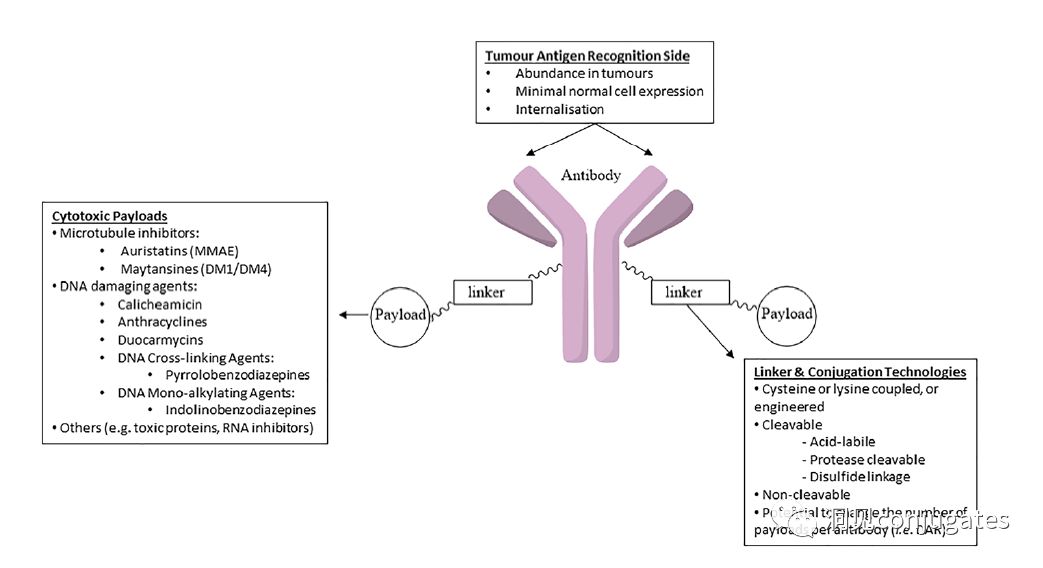
Antibody-drug conjugates (ADCs) consist of linkers, payloads, and monoclonal antibodies (mAbs). They combine the advantages of high specificity targeting and potent cytotoxic effects, achieving precise and efficient elimination of cancer cells, making them a hot topic in cancer drug development.
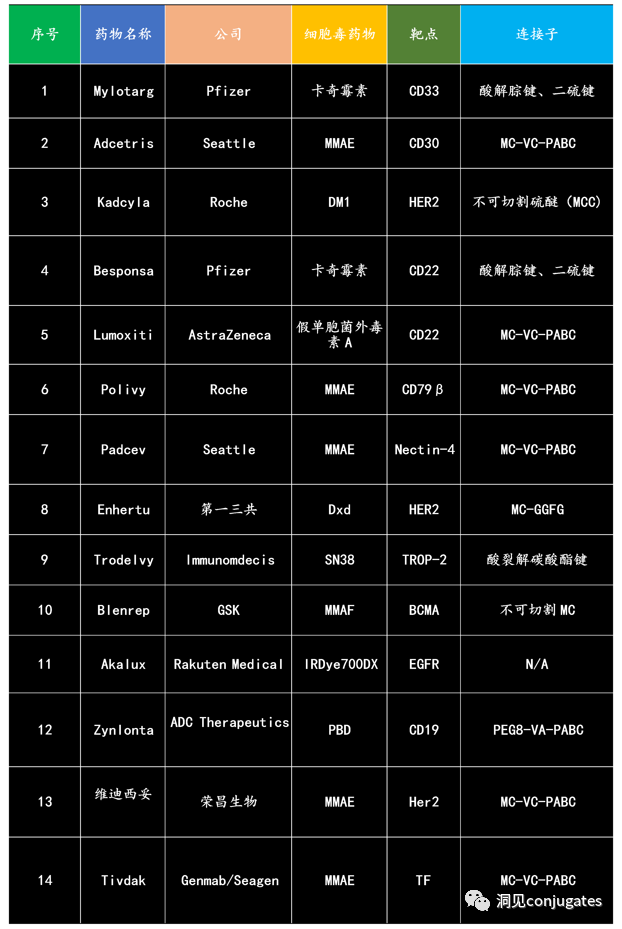
Since the first ADC drug Mylotarg® was approved by the FDA in 2000, as of December 2021, a total of 14 ADC drugs have been approved globally for hematological malignancies and solid tumors. Additionally, there are over 100 ADC candidates currently in various stages of clinical trials.
Antibody-drug conjugates (ADCs) consist of linkers, payloads, and monoclonal antibodies (mAbs). They combine the advantages of high specificity targeting and potent cytotoxic effects, achieving precise and efficient elimination of cancer cells, making them a hot topic in cancer drug development.
Since the first ADC drug Mylotarg® was approved by the FDA in 2000, as of December 2021, a total of 14 ADC drugs have been approved globally for hematological malignancies and solid tumors. Additionally, there are over 100 ADC candidates currently in various stages of clinical trials.
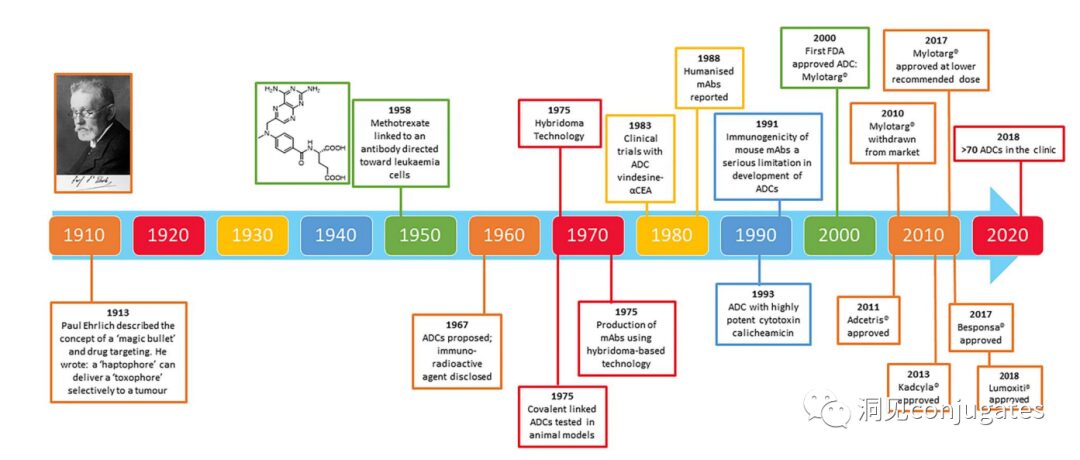
Timeline of Important Events in ADC Drug Development
As early as the early 20th century, Paul Ehrlich first proposed the concept of the “magic bullet” and hypothesized that certain compounds could directly enter tumor cells through specific targets, thereby curing diseases. Theoretically, these compounds should effectively kill cancer cells while being harmless to normal cells. In 2000, the U.S. Food and Drug Administration (FDA) approved the ADC drug Mylotarg® (gemtuzumab ozogamicin) for adult acute myeloid leukemia (AML), marking the beginning of the era of ADC targeted cancer therapy. Figure 1 depicts the iconic events in the development of ADC drugs from infancy to maturity over the past century. With the continuous expansion of targets and indications, ADCs are leading a new era of targeted cancer therapy, with the potential to replace traditional chemotherapy drugs in the future.
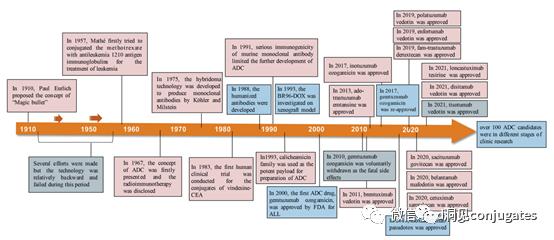 The concept of ADCs was initially described by Paul Ehrlich in the early 1900s, referring to them as “magic bullets.” Their development was not smooth, with significant progress not occurring until the early 1950s. During the 1980s and early 1990s, ADCs also faced many challenges.
Poor Target Antigen Selection is the most likely reason for early ADC failures, such as the KS1/4 antibody-methotrexate conjugate used for non-small cell lung cancer and the BR96 antibody-docetaxel conjugate used for metastatic breast cancer. Both of these ADCs entered clinical trials but provided almost no therapeutic benefit.
Other factors that may have limited the success of these early ADCs include their use of chimeric or murine antibodies that triggered immunogenic responses, as well as using payloads with low cytotoxic potency.
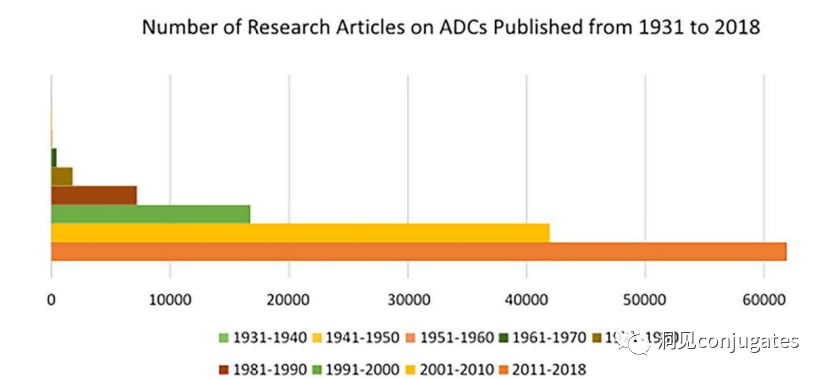
The field of ADC research has rapidly developed in recent years, as evidenced by the number of research papers published since 1970.
It is clear that the number of research articles doubled between 2000 and 2010, partly due to Kadcyla® and Adcetris® being in clinical development during this period. From 2011 to 2018, more than 60,000 research articles were published.
The concept of ADCs was initially described by Paul Ehrlich in the early 1900s, referring to them as “magic bullets.” Their development was not smooth, with significant progress not occurring until the early 1950s. During the 1980s and early 1990s, ADCs also faced many challenges.
Poor Target Antigen Selection is the most likely reason for early ADC failures, such as the KS1/4 antibody-methotrexate conjugate used for non-small cell lung cancer and the BR96 antibody-docetaxel conjugate used for metastatic breast cancer. Both of these ADCs entered clinical trials but provided almost no therapeutic benefit.
Other factors that may have limited the success of these early ADCs include their use of chimeric or murine antibodies that triggered immunogenic responses, as well as using payloads with low cytotoxic potency.
The field of ADC research has rapidly developed in recent years, as evidenced by the number of research papers published since 1970.
It is clear that the number of research articles doubled between 2000 and 2010, partly due to Kadcyla® and Adcetris® being in clinical development during this period. From 2011 to 2018, more than 60,000 research articles were published.
Mechanism of Action of ADCs
Traditional Internalization Mechanism
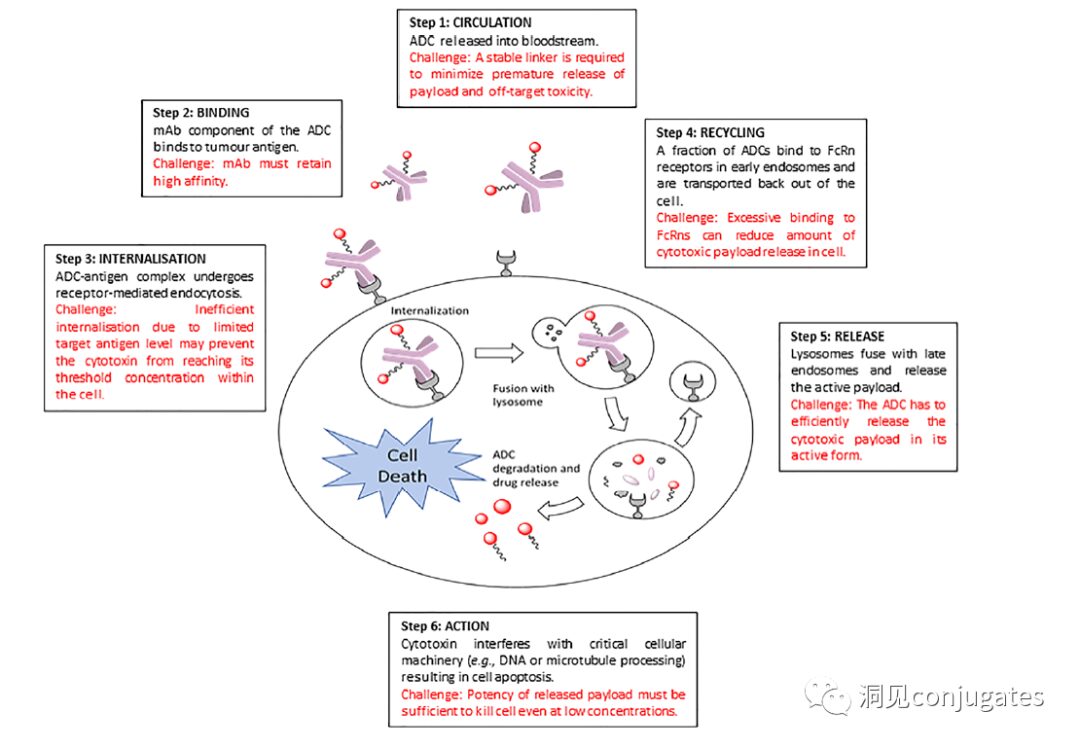
ADCs are linked via linkers and payloads to mAbs. ADCs bind to antigens on the surface of cancer cells and are then internalized, releasing highly cytotoxic payload molecules in the lysosome, typically through lysosomal cleavage.
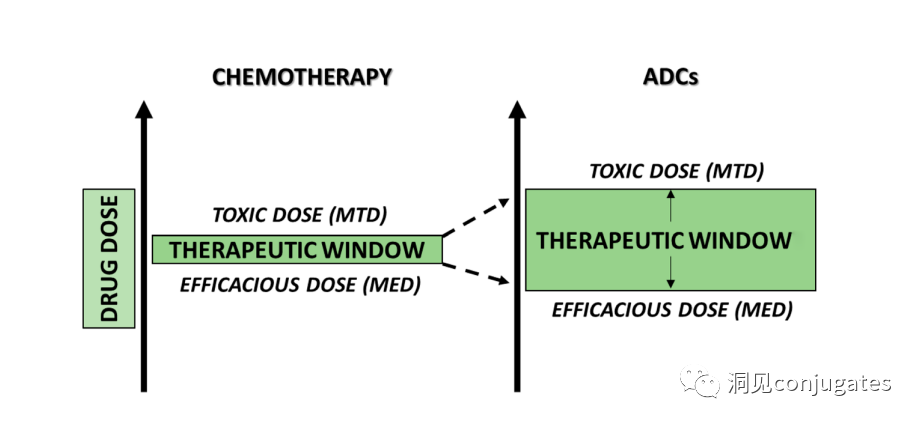
To design a successful ADC, it is crucial to understand the potential mechanisms of action. An effective ADC needs to retain the selectivity of the monoclonal antibody while being able to release a sufficiently high concentration of the payload to kill the target tumor cells. Each of these steps involves multiple unique challenges, making ADC design complex.
1: Prevent ADC from Releasing in Blood
2: ADC Must Bind to Tumor Antigen
3: Internalization of ADC-Antigen Complex
4: Recycling of ADC (Fc-mediated)
5: Lysosomal Release of Payload
6: Low Concentration Payload Kills Cancer Cells (Highly Toxic)
ADCs are designed to target and kill cancer cells, so antibodies must be able to recognize and bind to the corresponding antigens on tumor cells. Once bound to the antigen, the entire antigen-ADC complex is internalized through receptor-mediated endocytosis. The internalization process proceeds with the formation of early endosomes coated with clathrin containing the ADC-antigen complex. Once inside the lysosome, the ADC is degraded, and the cytotoxic payload is released into the cell, leading to cell death.
The mechanism of cell death will depend on the type of cytotoxic payload. For example, microtubule inhibitors, such as auristatins or maytansine, cause disruption of cytoplasmic division by interfering with microtubules; while DNA intercalators, such as PBD dimers or calicheamicin, cause DNA damage leading to apoptosis. New types of payloads under development will interfere with other cellular processes, such as RNA processing.
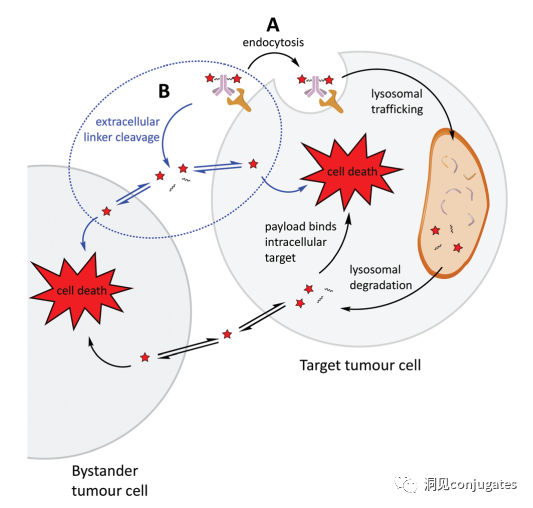
One important aspect of the ADC mechanism of action is the bystander effect, where free drugs exit tumor cells through the cell membrane into the tumor environment. This has potential therapeutic benefits in killing adjacent tumor cells, including those that may not express relevant antigens on their surface.
Another key factor is ensuring that a sufficient concentration of payload reaches the interior and kills cancer cells; in fact, this is a complex process that is difficult to guarantee. It is estimated that even if the overall mechanism of action of ADCs works at 50% efficiency, only 1-2% of the payload reaches the tumor cells. Therefore, it is important that the selected payload is sufficiently cytotoxic to be effective at very low concentrations. It is now recognized that the selection of antigen targets, antibodies, linkers, and payload components, as well as how they can effectively work together, is crucial for the success of ADCs.
Non-internalization Mechanism:
Linkers can break down in the extracellular tumor microenvironment to release the payload without requiring cellular endocytosis, allowing for non-internalizing antigens to be targeted.
One of the most critical factors in designing ADCs is the selection of antibodies. Most importantly, they must have high specificity for the antigen. Antibodies that lack high specificity and bind to other antigens may produce unpredictable effects, such as off-target toxicity due to interactions with healthy tissues or premature elimination before reaching the tumor site.
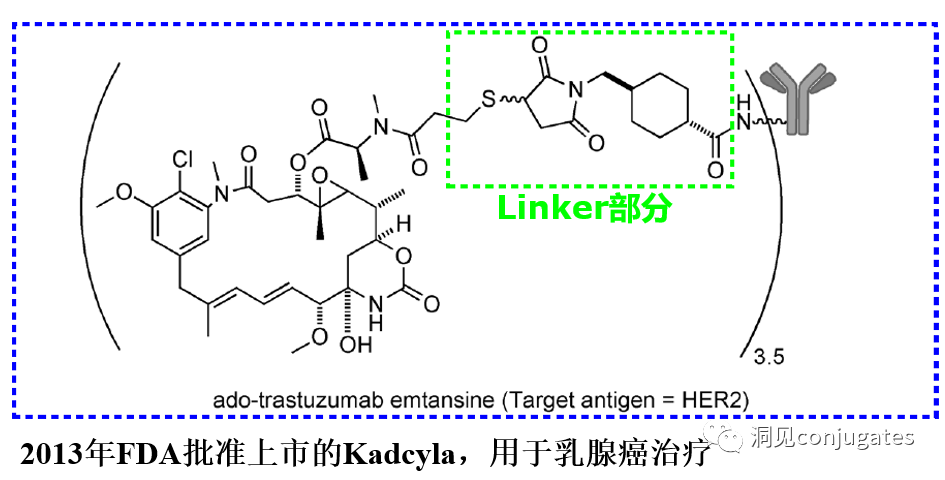
It is also important for antibodies to bind to target antigens with high affinity while having low immunogenicity. Another important characteristic is favorable pharmacokinetic (PK) properties. Finally, if the mAb has inherent anti-tumor activity by directly modulating the biological activity of the target antigen, this would be beneficial, as in the case of the anti-human epidermal growth factor receptor 2 (HER2) mAb trastuzumab (Herceptin®), which is the antibody component of trastuzumab emtansine (Kadcyla®).
Antibodies are classified into five categories based on the sequence of their heavy chain constant regions: Immunoglobulin M (IgM), IgD, IgG, IgE, and IgA.
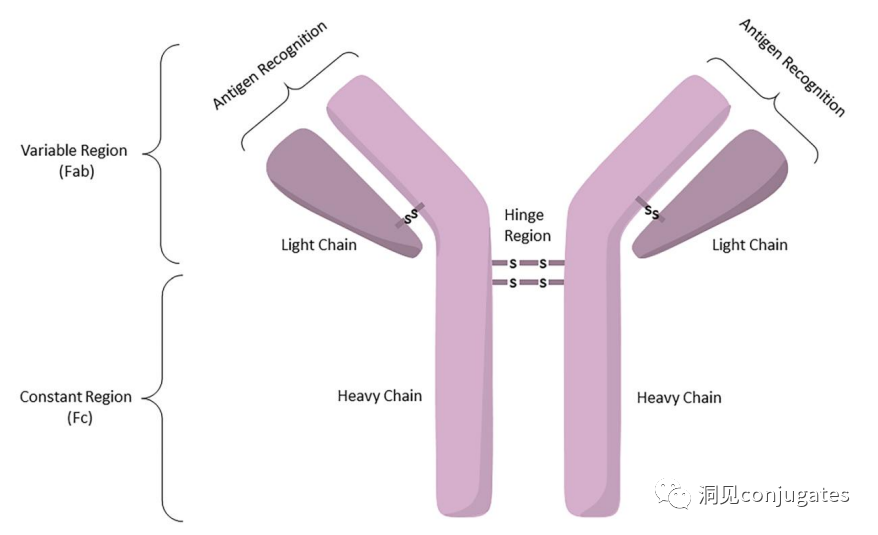
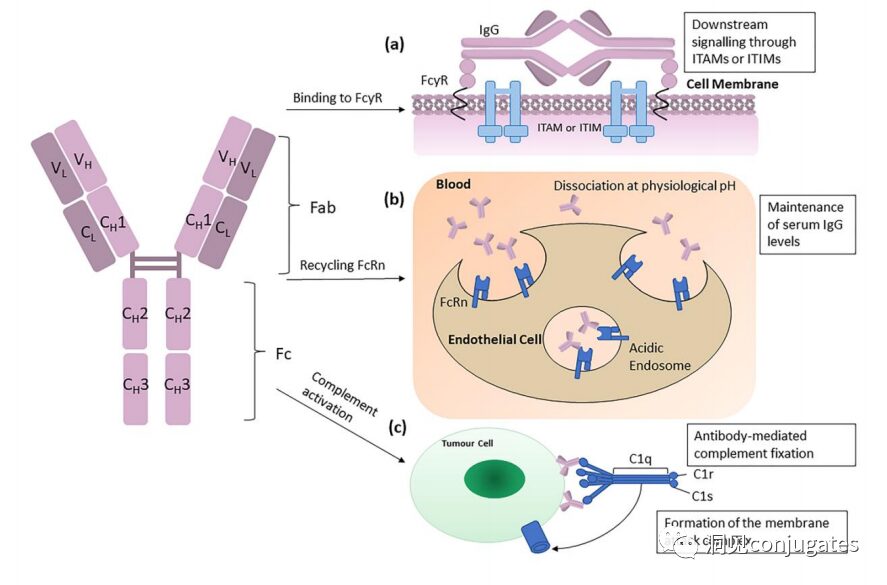
Among these five categories, IgG is the most commonly used in cancer immunotherapy. A typical IgG1 antibody consists of two heavy chains (H) and two light chains (L), including a constant (C) region that forms the Fc domain and a variable (V) region that provides antigen specificity in the Fab domain.
The Fc region is recognized by immune cells. The Fc fragment does not recognize the corresponding antigen but binds to various cell receptors (e.g., T cells) and complement proteins. All antibodies have glycosylation sites at conserved positions in their constant regions. For example, there is an N-glycosylation site at the N297 residue in the Fc region.
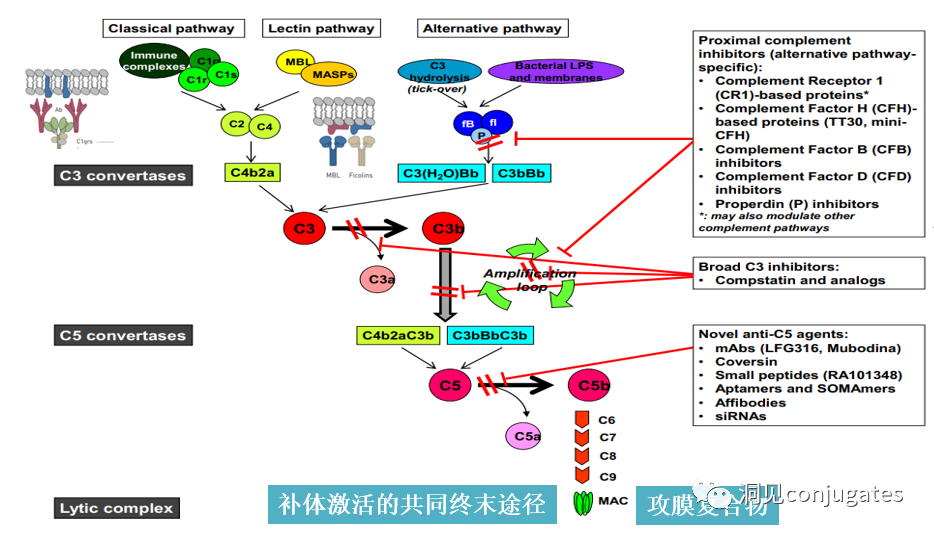
The subclasses of IgG, specifically IgG1 and IgG3, are effective activators of the classical complement pathway. Two or more IgG molecules bind to the complement component 1q (C1q) on the cell surface, with C1q binding to the Fc domain with high affinity, subsequently activating C1r enzyme activity, which then activates downstream complement proteins. The result of this cascade reaction is the formation of pores in the tumor cell surface by the membrane attack complex (MAC), leading to tumor cell lysis.
 To ensure effective internalization, the antigen binding site should have as high an affinity as possible (current research suggests that higher affinity is not always better; excessively high affinity may present issues in solid tumors). If the target is expressed abnormally in tumor tissues and the affinity is high, a large dose of ADC may be needed to saturate the available receptors at the tumor margins, allowing ADC molecules to penetrate deeper and bind. The only modifiable barrier effect of the binding site is the binding affinity; in the tumor microenvironment, antibody-drug conjugates are more likely to be captured by the first encountered target rather than penetrating to deeper locations. Therefore, optimizing affinity based on tumor size is essential to ensure sufficient affinity for drug internalization while allowing the drug to penetrate deeper into the tumor, necessitating a balance.
The following diagram illustrates how the IgG antibody antigen binds to Fc receptors and initiates signaling based on immunoreceptor tyrosine activation motifs (ITAM) or immunoreceptor tyrosine inhibitory motifs (ITIM). IgG can bind to Fc receptors (FcRn) on endothelial cells to maintain serum IgG levels and bind to tumor cells, where they can recruit complement component 1q (C1q) to initiate the complement cascade, leading to the lysis of tumor cells.
Considering the high specificity, affinity, and long exposure time required at the tumor site, ideally, antibody selection should ensure minimal interaction with healthy tissues, sub-nanomolar affinity for target antigens, and a longer pharmacokinetic half-life, with minimal immunogenicity.
The degree of immunogenicity of ADCs is a key factor and an important determinant of the circulation half-life. In particular, antibodies that lack tumor specificity may be rapidly cleared from circulation due to immunogenicity, leading to reduced therapeutic efficacy. In the early development of ADCs, studies were based on murine monoclonal antibodies, resulting in the formation of human anti-mouse antibodies within weeks after a single administration.
As a result, mouse antibodies were quickly replaced by chimeric IgG antibodies, followed by humanized IgG. In recent years, ADCs have mainly been based on fully human antibodies. One of the most significant advantages of using mAbs rather than small molecular chemotherapeutics for cancer treatment is that mAb-based drugs can have favorable pharmacokinetics in terms of duration, metabolism, and elimination.
As mentioned above, once mAbs are administered into the bloodstream, they can distribute to tumor tissues either through extravasation via pores in the endothelium or through pinocytosis. The distribution of ADCs in tumor tissues is limited by the size of the antibodies, which typically accounts for 95% of the ADC mass. However, unlike normal blood vessels, which have tightly connected endothelial cells in a monolayer, tumor endothelium is often characterized by excessive branching and sprouting, leading to “leaky” monolayers. Thus, although ADCs are larger molecules, they may still distribute to tumor tissues through the leaky vascular system, but their distribution in metabolic and clearance organs such as the liver, intestines, muscles, and skin is limited. Recently, antibody fragments are being investigated; due to their smaller size, they may penetrate tumor tissues better compared to full-size antibodies, which typically have shorter half-lives.
The design, structure, and chemical properties of linkers are crucial for the characteristics of ADCs, affecting the specificity, efficacy, and safety of the entire ADC drug. Generally, linkers are designed to be stable in the bloodstream while effectively releasing the payload at the tumor site. It is also critical that the conjugate remains stable in buffered aqueous solutions. Linkers are categorized based on their payload release mechanisms, primarily into two types: “cleavable” or “non-cleavable” (Recommended reading: Common types of linkers in ADC drugs and their in vivo cleavage mechanisms).
To ensure effective internalization, the antigen binding site should have as high an affinity as possible (current research suggests that higher affinity is not always better; excessively high affinity may present issues in solid tumors). If the target is expressed abnormally in tumor tissues and the affinity is high, a large dose of ADC may be needed to saturate the available receptors at the tumor margins, allowing ADC molecules to penetrate deeper and bind. The only modifiable barrier effect of the binding site is the binding affinity; in the tumor microenvironment, antibody-drug conjugates are more likely to be captured by the first encountered target rather than penetrating to deeper locations. Therefore, optimizing affinity based on tumor size is essential to ensure sufficient affinity for drug internalization while allowing the drug to penetrate deeper into the tumor, necessitating a balance.
The following diagram illustrates how the IgG antibody antigen binds to Fc receptors and initiates signaling based on immunoreceptor tyrosine activation motifs (ITAM) or immunoreceptor tyrosine inhibitory motifs (ITIM). IgG can bind to Fc receptors (FcRn) on endothelial cells to maintain serum IgG levels and bind to tumor cells, where they can recruit complement component 1q (C1q) to initiate the complement cascade, leading to the lysis of tumor cells.
Considering the high specificity, affinity, and long exposure time required at the tumor site, ideally, antibody selection should ensure minimal interaction with healthy tissues, sub-nanomolar affinity for target antigens, and a longer pharmacokinetic half-life, with minimal immunogenicity.
The degree of immunogenicity of ADCs is a key factor and an important determinant of the circulation half-life. In particular, antibodies that lack tumor specificity may be rapidly cleared from circulation due to immunogenicity, leading to reduced therapeutic efficacy. In the early development of ADCs, studies were based on murine monoclonal antibodies, resulting in the formation of human anti-mouse antibodies within weeks after a single administration.
As a result, mouse antibodies were quickly replaced by chimeric IgG antibodies, followed by humanized IgG. In recent years, ADCs have mainly been based on fully human antibodies. One of the most significant advantages of using mAbs rather than small molecular chemotherapeutics for cancer treatment is that mAb-based drugs can have favorable pharmacokinetics in terms of duration, metabolism, and elimination.
As mentioned above, once mAbs are administered into the bloodstream, they can distribute to tumor tissues either through extravasation via pores in the endothelium or through pinocytosis. The distribution of ADCs in tumor tissues is limited by the size of the antibodies, which typically accounts for 95% of the ADC mass. However, unlike normal blood vessels, which have tightly connected endothelial cells in a monolayer, tumor endothelium is often characterized by excessive branching and sprouting, leading to “leaky” monolayers. Thus, although ADCs are larger molecules, they may still distribute to tumor tissues through the leaky vascular system, but their distribution in metabolic and clearance organs such as the liver, intestines, muscles, and skin is limited. Recently, antibody fragments are being investigated; due to their smaller size, they may penetrate tumor tissues better compared to full-size antibodies, which typically have shorter half-lives.
The design, structure, and chemical properties of linkers are crucial for the characteristics of ADCs, affecting the specificity, efficacy, and safety of the entire ADC drug. Generally, linkers are designed to be stable in the bloodstream while effectively releasing the payload at the tumor site. It is also critical that the conjugate remains stable in buffered aqueous solutions. Linkers are categorized based on their payload release mechanisms, primarily into two types: “cleavable” or “non-cleavable” (Recommended reading: Common types of linkers in ADC drugs and their in vivo cleavage mechanisms).
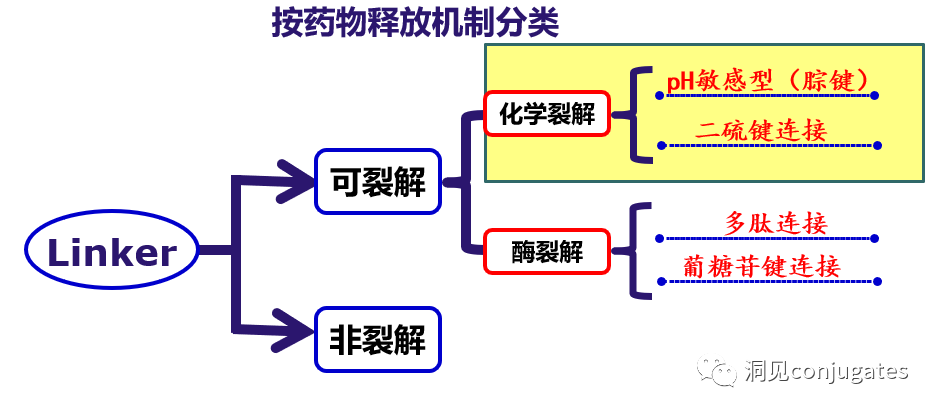
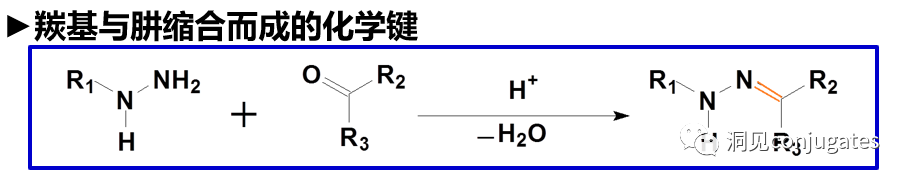
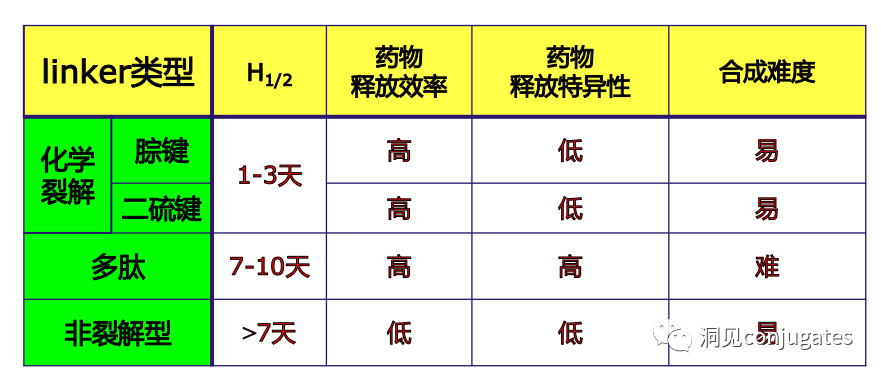 Cleavable linkers utilize the differences in conditions between the bloodstream and the intracellular cytoplasm of tumor cells. Changes in the environment after internalization of the ADC-antigen complex trigger the cleavage of the linker and the release of the active payload. Cleavable linkers are divided into three main subclasses: (1) acid-labile (e.g., hydrazone), (2) reducible (e.g., disulfide), and (3) enzyme-labile (e.g., peptide). The diagram below shows an example of a hydrazone linker used in Gemtuzumab ozogamicin (Mylotarg®). Non-specific release of the payload may lead to systemic toxicity. This is also one of the reasons Mylotarg was withdrawn from the market in 2010.
Cleavable linkers utilize the differences in conditions between the bloodstream and the intracellular cytoplasm of tumor cells. Changes in the environment after internalization of the ADC-antigen complex trigger the cleavage of the linker and the release of the active payload. Cleavable linkers are divided into three main subclasses: (1) acid-labile (e.g., hydrazone), (2) reducible (e.g., disulfide), and (3) enzyme-labile (e.g., peptide). The diagram below shows an example of a hydrazone linker used in Gemtuzumab ozogamicin (Mylotarg®). Non-specific release of the payload may lead to systemic toxicity. This is also one of the reasons Mylotarg was withdrawn from the market in 2010.
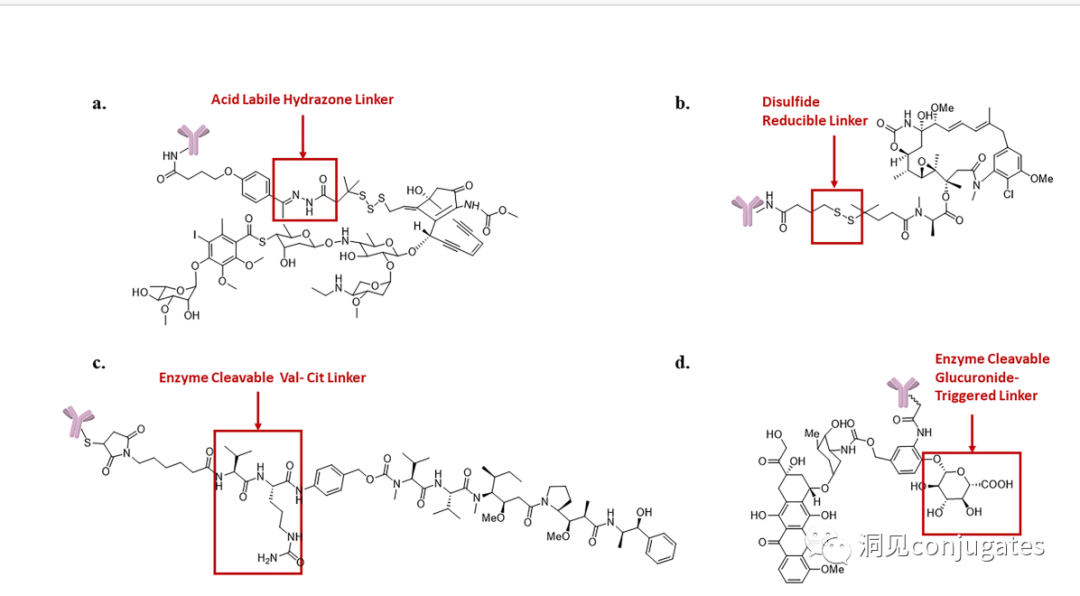 Another type of enzyme-cleavable linker is based on β-glucuronide. β-glucuronidase (in lysosomes) can release the payload from linkers containing β-glucuronide and is overexpressed in certain tumor cell types. An important feature of β-glucuronide linkers is their hydrophilicity, which can potentially reduce aggregation during the conjugation process compared to structures based on dipeptides or other linker types. β-glucuronide has been used as a linker component to connect payloads, including auristatin derivatives MMAE and MMAF, as well as doxorubicin.
Chemical Cleavable Linkers:
In a neutral blood environment (pH 7.3-7.5): about 6% cleavage rate; in the intracellular lysosome (pH 4.5-5.0) or endosomes (pH 5.0-6.5), it cleaves easily, with a cleavage rate of about 97% at pH 4.5.
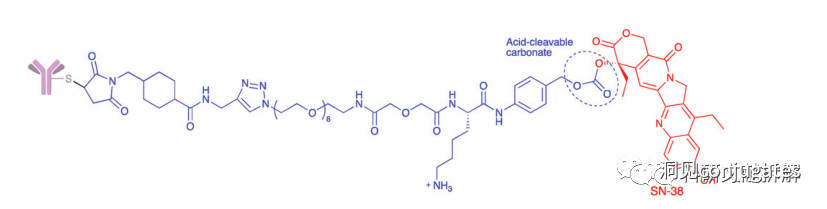
Another commonly used structure for acid condition hydrolysis is carbonate linkers.
Simple carbonates have limited stability in serum and require the addition of an amino benzyl (PAB) to significantly improve half-life.
Carbonate structures release SN-38 drugs and carbon dioxide under acidic conditions.
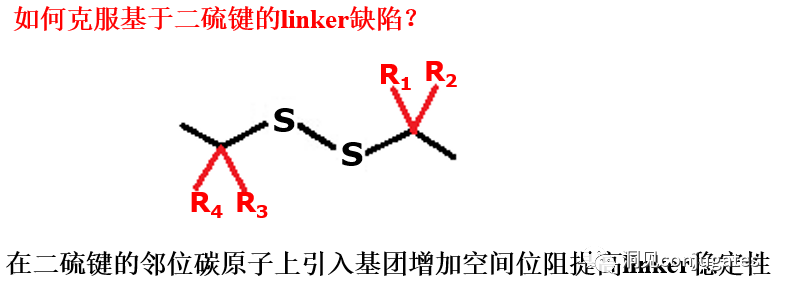
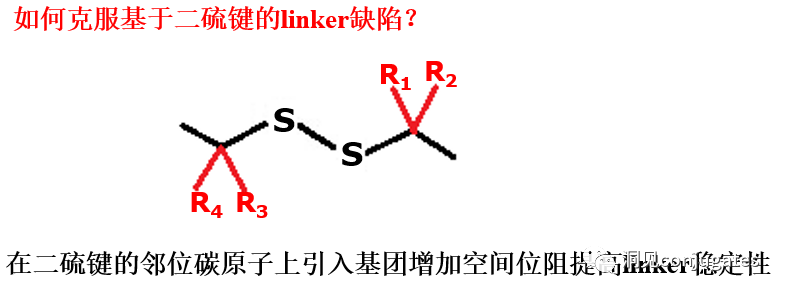
2: Disulfide Chemical Cleavage
Disulfide compounds are stable under physiological pH conditions but are easily attacked by nucleophiles from thiols, breaking the link (including sterically hindered disulfide bonds).
There are complex thiol-containing compounds in the blood, along with interference from albumin, which contains free thiol small molecules (GSH and Cys):
In the blood environment: 0.005 mM
In the intracellular environment: 0.5 -10 mM
The average concentration of GSH and Cys in cancer cells is even higher
Enzyme Cleavable Linkers:
Specific enzymes are present in both the intracellular lysosomes and the extracellular tumor microenvironment, which can selectively cleave the corresponding substrates. Peptide-based linkers utilize proteolytic enzymes such as Cathepsin and plasmin in lysosomes (these proteolytic enzymes are strictly inhibited in the blood environment, while enzyme activity is optimal in the lysosomal environment).
►Proteolytic enzymes are generally expressed at higher levels in cancer cells
►Peptide-based linkers have higher stability in blood and cleave faster in cancer cells
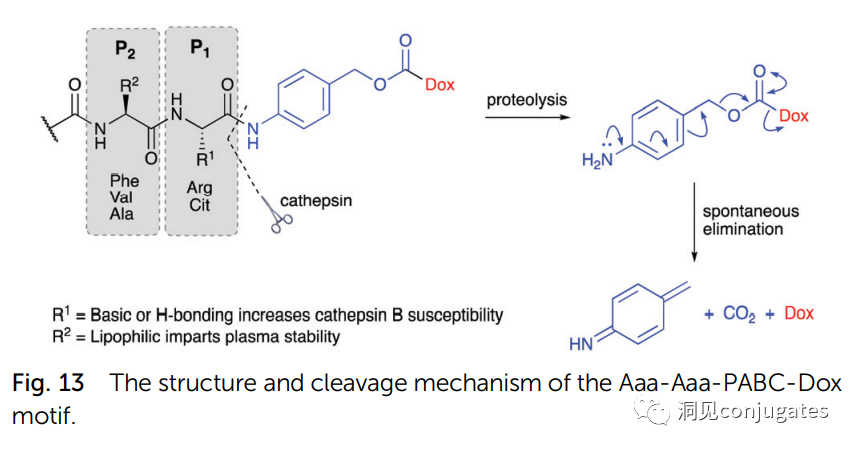
1: Cathepsin B Cleavage of Dipeptides:
Linkers Based on Dipeptides + Self-Cleaving Modules:
The best dipeptides: Val-Cit and Phe-Lys (citrulline-valine and phenylalanine-lysine), can be hydrolyzed by Cathepsin B-specific enzymes.
Self-Cleaving Module: PABC
Dipeptide linkers are typically targeted at Cathepsin B, which is primarily present in lysosomes, although activity is also observed extracellularly.
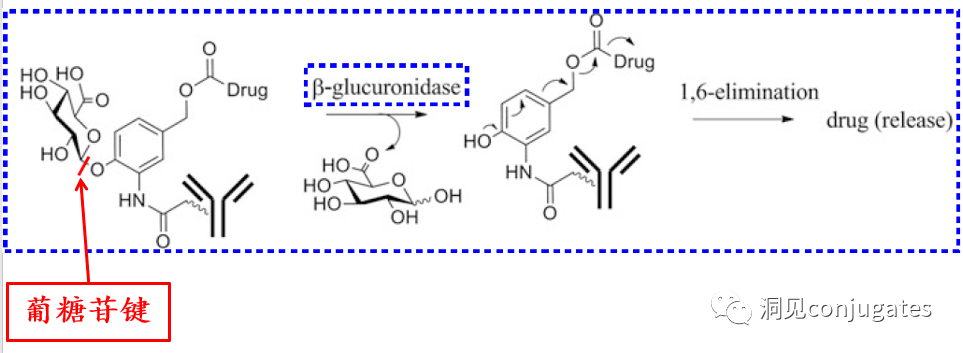
β-glucuronidase is a hydrolytic enzyme among glycosidases, catalyzing the breakdown of β-glucuronic acid residues in polysaccharides using β-glucuronidase in lysosomes.
►β-glucuronidase is abundant in lysosomes and is overexpressed in certain cancer cells.
►The activity of β-glucuronidase in the lysosomal environment is highest, while enzyme activity is inhibited in the blood environment.
Glucuronide linkers have another advantage; due to their high hydrophilicity, they can significantly reduce the aggregation of ADCs, allowing for the synthesis of ADC drugs with high payloads (DAR=8).
β-galactosidase can cleave linkers similarly to glucuronidase.
Acidic pyrophosphatases and acid phosphatases present in lysosomes can hydrolyze phosphate ester bonds.
Another type of enzyme-cleavable linker is based on β-glucuronide. β-glucuronidase (in lysosomes) can release the payload from linkers containing β-glucuronide and is overexpressed in certain tumor cell types. An important feature of β-glucuronide linkers is their hydrophilicity, which can potentially reduce aggregation during the conjugation process compared to structures based on dipeptides or other linker types. β-glucuronide has been used as a linker component to connect payloads, including auristatin derivatives MMAE and MMAF, as well as doxorubicin.
Chemical Cleavable Linkers:
In a neutral blood environment (pH 7.3-7.5): about 6% cleavage rate; in the intracellular lysosome (pH 4.5-5.0) or endosomes (pH 5.0-6.5), it cleaves easily, with a cleavage rate of about 97% at pH 4.5.
Another commonly used structure for acid condition hydrolysis is carbonate linkers.
Simple carbonates have limited stability in serum and require the addition of an amino benzyl (PAB) to significantly improve half-life.
Carbonate structures release SN-38 drugs and carbon dioxide under acidic conditions.
2: Disulfide Chemical Cleavage
Disulfide compounds are stable under physiological pH conditions but are easily attacked by nucleophiles from thiols, breaking the link (including sterically hindered disulfide bonds).
There are complex thiol-containing compounds in the blood, along with interference from albumin, which contains free thiol small molecules (GSH and Cys):
In the blood environment: 0.005 mM
In the intracellular environment: 0.5 -10 mM
The average concentration of GSH and Cys in cancer cells is even higher
Enzyme Cleavable Linkers:
Specific enzymes are present in both the intracellular lysosomes and the extracellular tumor microenvironment, which can selectively cleave the corresponding substrates. Peptide-based linkers utilize proteolytic enzymes such as Cathepsin and plasmin in lysosomes (these proteolytic enzymes are strictly inhibited in the blood environment, while enzyme activity is optimal in the lysosomal environment).
►Proteolytic enzymes are generally expressed at higher levels in cancer cells
►Peptide-based linkers have higher stability in blood and cleave faster in cancer cells
1: Cathepsin B Cleavage of Dipeptides:
Linkers Based on Dipeptides + Self-Cleaving Modules:
The best dipeptides: Val-Cit and Phe-Lys (citrulline-valine and phenylalanine-lysine), can be hydrolyzed by Cathepsin B-specific enzymes.
Self-Cleaving Module: PABC
Dipeptide linkers are typically targeted at Cathepsin B, which is primarily present in lysosomes, although activity is also observed extracellularly.
β-glucuronidase is a hydrolytic enzyme among glycosidases, catalyzing the breakdown of β-glucuronic acid residues in polysaccharides using β-glucuronidase in lysosomes.
►β-glucuronidase is abundant in lysosomes and is overexpressed in certain cancer cells.
►The activity of β-glucuronidase in the lysosomal environment is highest, while enzyme activity is inhibited in the blood environment.
Glucuronide linkers have another advantage; due to their high hydrophilicity, they can significantly reduce the aggregation of ADCs, allowing for the synthesis of ADC drugs with high payloads (DAR=8).
β-galactosidase can cleave linkers similarly to glucuronidase.
Acidic pyrophosphatases and acid phosphatases present in lysosomes can hydrolyze phosphate ester bonds.
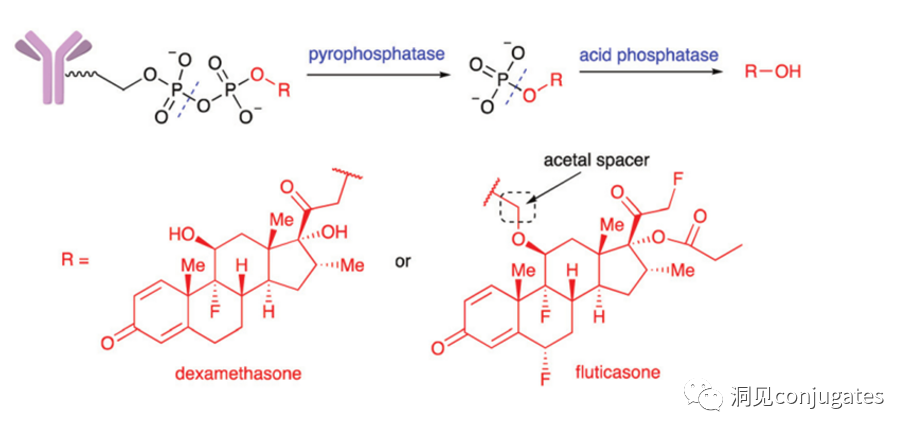 Merck designed a diphosphate linker that releases prototype glucocorticoid payloads upon hydrolysis in lysosomes; when testing a monophosphate linker, drug release was relatively slowed.
►ADCs containing non-cleavable linkers:
Once ADCs are phagocytosed into the cell, they enter the lysosomal pathway, where, under the action of various cleavage enzymes, the drug derivative is released after complete degradation of the antibody, killing the target cells. Generally, they have better stability.
►Requirements for Non-Cleavable Linkers:
(1) Linkers must not have degradation conditions and factors outside the cell.
(2) The drug derivatives released after intracellular degradation of ADCs should also have considerable cytotoxicity, so the use of these linkers has strict requirements for the drugs.
►Disadvantages of Non-Cleavable Linkers:
(1) Strict requirements for drugs, necessitating screening and validation of the cytotoxicity of drug derivatives.
(2) The drug release rate is relatively slow.
(3) Generally, drug derivatives after intracellular degradation of ADCs have good water solubility, and may even carry charged groups, which is not conducive to exerting the bystander effect (Bystander Kill Effect).
Merck designed a diphosphate linker that releases prototype glucocorticoid payloads upon hydrolysis in lysosomes; when testing a monophosphate linker, drug release was relatively slowed.
►ADCs containing non-cleavable linkers:
Once ADCs are phagocytosed into the cell, they enter the lysosomal pathway, where, under the action of various cleavage enzymes, the drug derivative is released after complete degradation of the antibody, killing the target cells. Generally, they have better stability.
►Requirements for Non-Cleavable Linkers:
(1) Linkers must not have degradation conditions and factors outside the cell.
(2) The drug derivatives released after intracellular degradation of ADCs should also have considerable cytotoxicity, so the use of these linkers has strict requirements for the drugs.
►Disadvantages of Non-Cleavable Linkers:
(1) Strict requirements for drugs, necessitating screening and validation of the cytotoxicity of drug derivatives.
(2) The drug release rate is relatively slow.
(3) Generally, drug derivatives after intracellular degradation of ADCs have good water solubility, and may even carry charged groups, which is not conducive to exerting the bystander effect (Bystander Kill Effect).
References:
1. Cytotoxic payloads for antibody-drug conjugates
2. Other publicly available information compiled
This course invited Ms. Ye Feng, head of cell line development at Kangri Baiao, to deliver a wonderful lecture on “Analysis of Bispecific Antibody Cell Line Construction Strategies” for colleagues interested in the field of cell line development.
June 22, 2022 (Wednesday)
19:30-20:30
Theme:Analysis of Bispecific Antibody Cell Line Construction Strategies
Live Broadcast Content:
-
The development history of bispecific antibodies
-
Advantages and disadvantages of different bispecific antibody configurations
-
Development strategies for bispecific antibody cell lines
Speaker:

-
Previously responsible for cell line construction work at several well-known pharmaceutical companies, with rich experience in cell line development, familiar with various host cell development platforms such as CHO-K1 (ATCC), CHOZN-K1, CHO-S, and DG44, having participated in the cell line development of over 20 projects involving mAbs, bispecific antibodies, cytokines, fusion proteins, and ADCs, with multiple projects entering clinical stages. Established and optimized the cell line construction platform at Kangri Baiao, successfully delivering nearly 30 projects.
Scan the QR code below to directly enter the live broadcast room.

Copy the following link and open it in your browser to participate.
Web link: https://hso.h5.xeknow.com/sl/3gGlLb
Kangri Baiao Biotechnology (Suzhou) Co., Ltd. is a professional biopharmaceutical CDMO. The company’s services include cell line development, bulk and formulation process development, analytical method development, formulation development, and cGMP production of bulk and finished products. The team members are all experienced senior professionals in the biopharmaceutical industry, with a profound understanding of the CMC field of biopharmaceuticals. Their extensive experience accumulated in leading biopharmaceutical R&D and manufacturing companies is key to Kangri Baiao’s rapid establishment and provision of high-quality services to clients. Kangri Baiao has successfully assisted multiple partner projects in obtaining clinical approvals in China, the United States, Australia, Europe, and other regions.
Kangri Baiao is committed to providing efficient and cost-effective biopharmaceutical outsourcing service solutions for global partners, helping partners shorten the time to enter clinical trials and market. For more information, please follow the official website of Kangri Baiao: www.bioworkshops.com or call 0512-67999700.
Scan the WeChat QR code to add the Antibody Circle editor, and eligible individuals can join the Antibody Circle WeChat group!
Please indicate: Name + Research Direction!

All articles reproduced in this public account are for the purpose of conveying more information, and the source and author are clearly indicated. Media or individuals wishing not to be reproduced can contact us ([email protected]), and we will immediately delete them. All articles represent the author’s views and do not represent the position of this site.