Research Background
Microorganisms are the most numerous and widely distributed forms of life on Earth, playing an indispensable role in biogeochemical cycles and ecological balance. With the rapid development of high-throughput sequencing technology, metagenomics has become a powerful tool for studying the structure and function of microbial communities. Although traditional 16S rRNA amplicon sequencing can provide an overview of community composition, it has limited capacity for analysis at the species or even strain level. In contrast, shotgun metagenomic sequencing allows for direct sequencing of all DNA in environmental samples, making it possible to reconstruct the genomes of individual microorganisms—known as Metagenome-Assembled Genomes (MAGs). However, over 99% of microorganisms in nature are difficult to culture in the laboratory, constituting a vast “microbial dark matter.” Through culture-independent MAG reconstruction techniques, scientists have gained insights into the genetic information of this unknown microbial diversity, greatly expanding our understanding of the tree of life. Despite the numerous MAGs reported in recent years, their reconstruction process is computationally intensive, time-consuming, and lacks a centralized, rigorously quality-controlled, and standardized high-quality MAG database, which limits data reuse and cross-study comparisons.

Paper Summary
The team led by Tao Li from Zhejiang University School of Medicine collaborated with the team of Ting-Ting Li from Nanhu Laboratory to publish a paper titled “MAGdb: a comprehensive high quality MAGs repository for exploring microbial metagenome-assemble genomes” in Genome Biology. They constructed a comprehensive high-quality metagenome-assembled genomes (MAGs) database named MAGdb. This database integrates clinical, environmental, and animal metagenomic data from around the world, reconstructing nearly 100,000 high-quality MAGs through standardized analytical processes, and provides a user-friendly online platform aimed at offering researchers a convenient and standardized resource to explore microbial diversity, functional potential, and ecological roles.
Main Research Results
Construction and Core Content of MAGdb
The construction of MAGdb follows a rigorous three-phase workflow (Figure 1).The first phase is data collection and organization: The research team systematically screened over 2,000 metagenomic studies published since 2015, ultimately selecting 74 representative studies covering clinical, environmental, and animal domains. They downloaded 13,702 raw metagenomic sequencing data (runs) related to these studies from public databases and manually organized detailed metadata, including sample sources, geographic locations, host information, etc., providing a solid foundation for subsequent analyses.
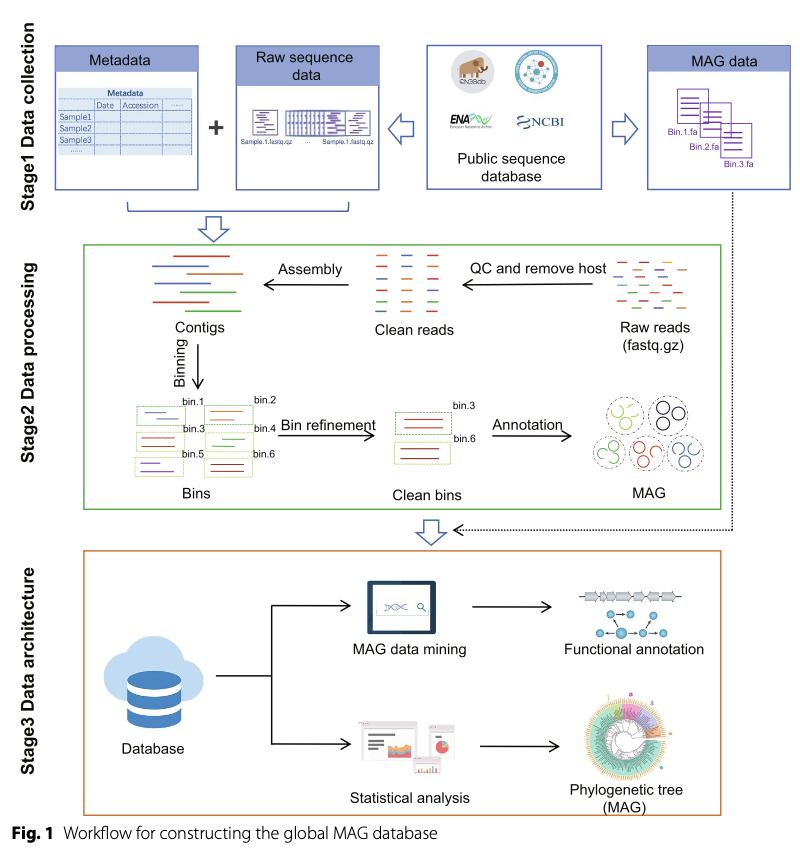
The second phase is standardized data processing and MAG reconstruction: To ensure data consistency and comparability, the team applied a standardized bioinformatics analysis workflow to all raw sequencing data that did not provide MAGs. This workflow includes raw data quality control, removal of host genome contamination, metagenomic assembly, genome binning, and optimization. Specifically, to improve the accuracy and completeness of binning, they integrated three mainstream binning tools and optimized and deduplicated them using the metaWRAP tool. Finally, CheckM software was used to assess the quality of all reconstructed MAGs, retaining only those with completeness greater than 90% and contamination less than 5% as high-quality MAGs (HMAGs) for inclusion in the database.
The third phase is database architecture and web platform implementation: All processed HMAG sequences, species annotations, functional annotations, and related metadata were integrated into a MongoDB database. The team developed an interactive web interface to facilitate data browsing, searching, and downloading for users.
Database Statistics and Overview of Microbial Diversity
As of the publication of the paper, MAGdb has recorded 99,672 HMAGs from 66 countries across five continents. These data primarily originate from clinical samples (76.2%), followed by environmental samples (12.04%) and animal samples (11.4%) (Figure 2a, Table 1). All HMAGs included in the database meet or exceed the high-quality standards of MIMAG (Minimum Information about Metagenome-Assembled Genomes), with an average completeness of 96.84% and an average contamination of only 1.02%, reflecting the genetic characteristics of different microbial groups (Figure 2b).
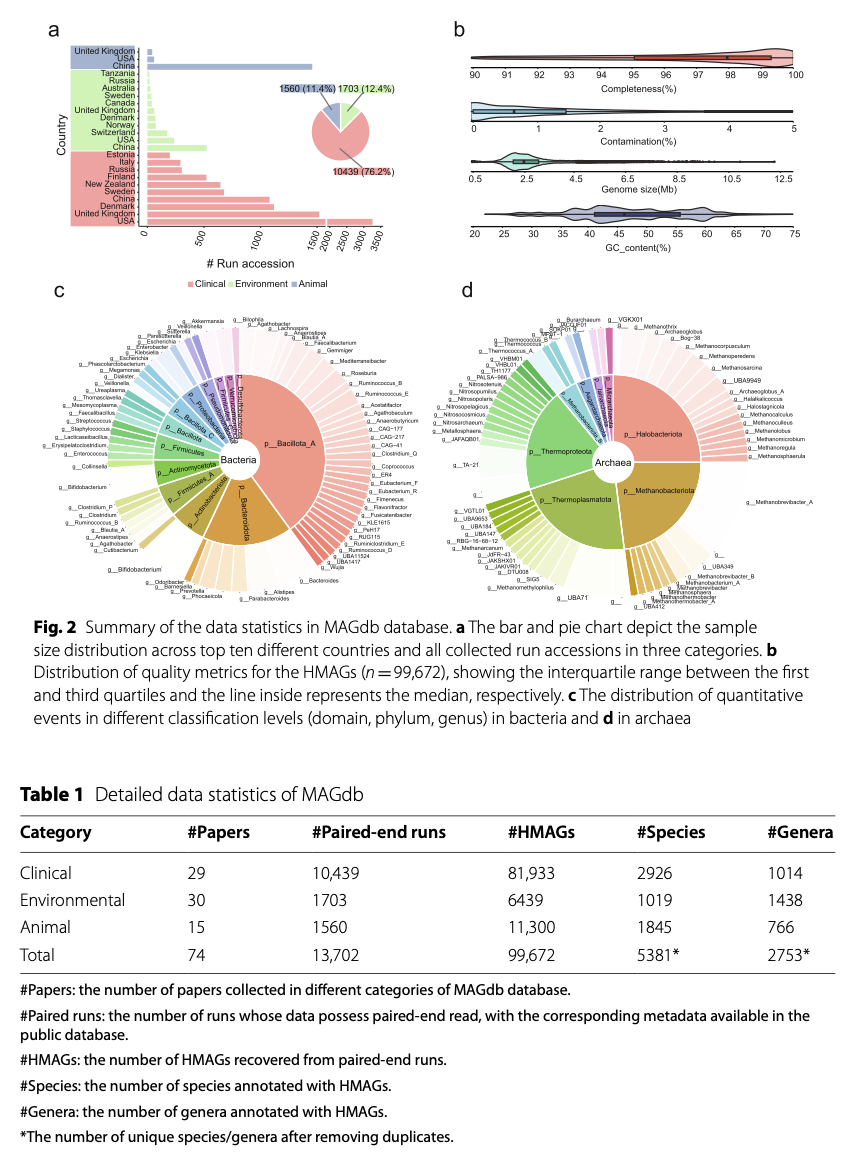
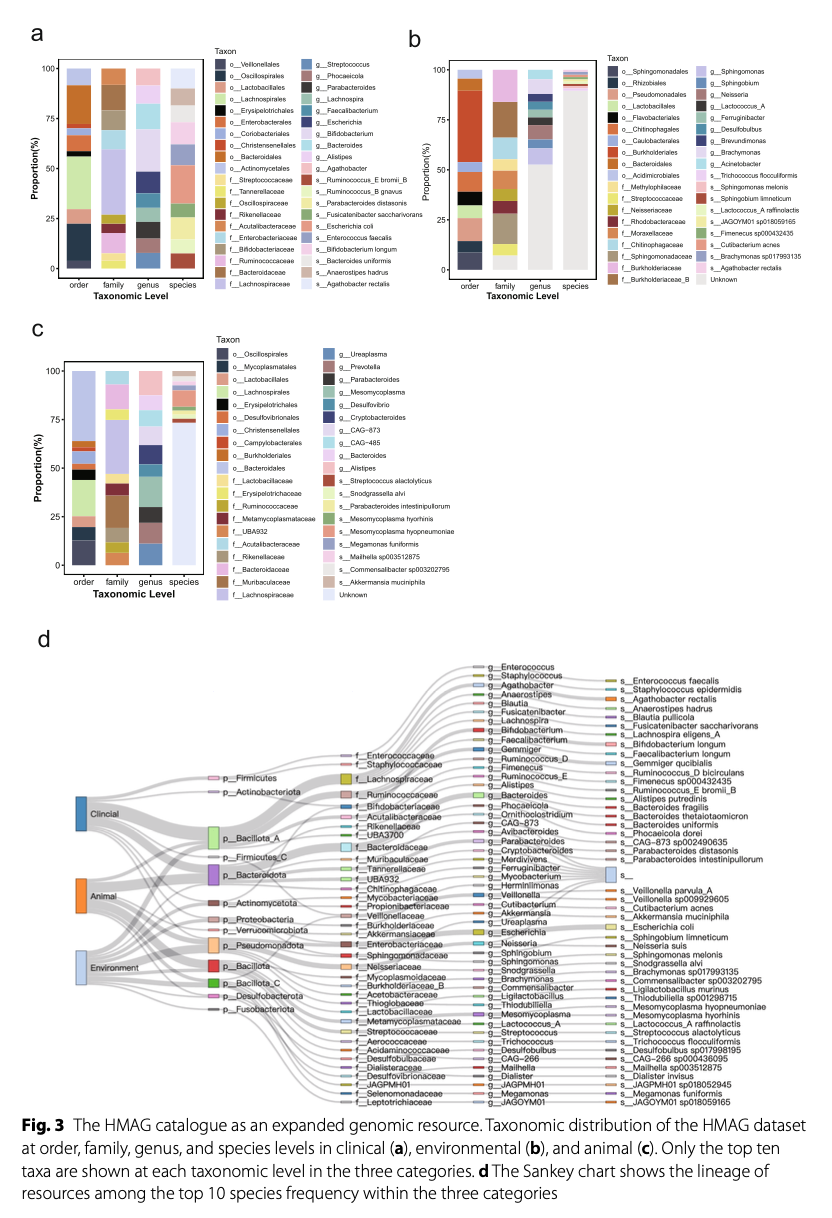
The species annotation results reveal that MAGdb contains a vast microbial diversity. These HMAGs cover 90 known phyla (82 bacterial phyla and 8 archaeal phyla), identifying a total of 5,381 different species and 2,753 genera. Notably, 6.3% (6,316) of HMAGs remain unannotated at the species level, indicating that MAGdb contains a significant number of potential new species, making it a valuable resource for exploring microbial “dark matter.” Among different sample types, species distribution also shows significant differences: clinical samples are dominated by well-known species such as Escherichia coli, while environmental and animal samples contain a large number of unclassified species, further highlighting the unexplored microbial diversity in these ecosystems (Figure 3).
User-Friendly Interactive Interface and Functional Modules
MAGdb provides a cleanly designed, powerful web platform, primarily consisting of three core modules: “Rawdata,” “MAG,” and “Download” (Figure 4).
- Rawdata Module: Users can browse all raw datasets by research paper. Each entry provides links to the original literature, sample counts, quality control reports, and detailed metadata downloads. This design ensures complete traceability of the data, allowing users to clearly understand the source of each HMAG.
- MAG Module: This is the core browsing module of the database. Users can view a list of all MAGs generated in each study and click to enter the detail page of each HMAG. The detail page presents quality statistics (such as completeness, contamination, N50, etc.) and species classification information in graphical form.
- Download Module: This module provides users with convenient data download functionality. Users can batch download HMAG genome sequence files by research project or select multiple projects for one-time download.

Phylogenetic and Functional Exploration Based on MAGdb
To demonstrate the scientific value of MAGdb, the research team conducted in-depth exploratory analyses of the database content. They first constructed a phylogenetic tree containing 7,303 representative HMAGs (deduplicated at 95% average nucleotide identity level). The results showed that these genomes are widely distributed on the tree of life, particularly some bacterial phyla (e.g., p_Bacillota_A) exhibiting extremely high phylogenetic diversity (Figure 5a).
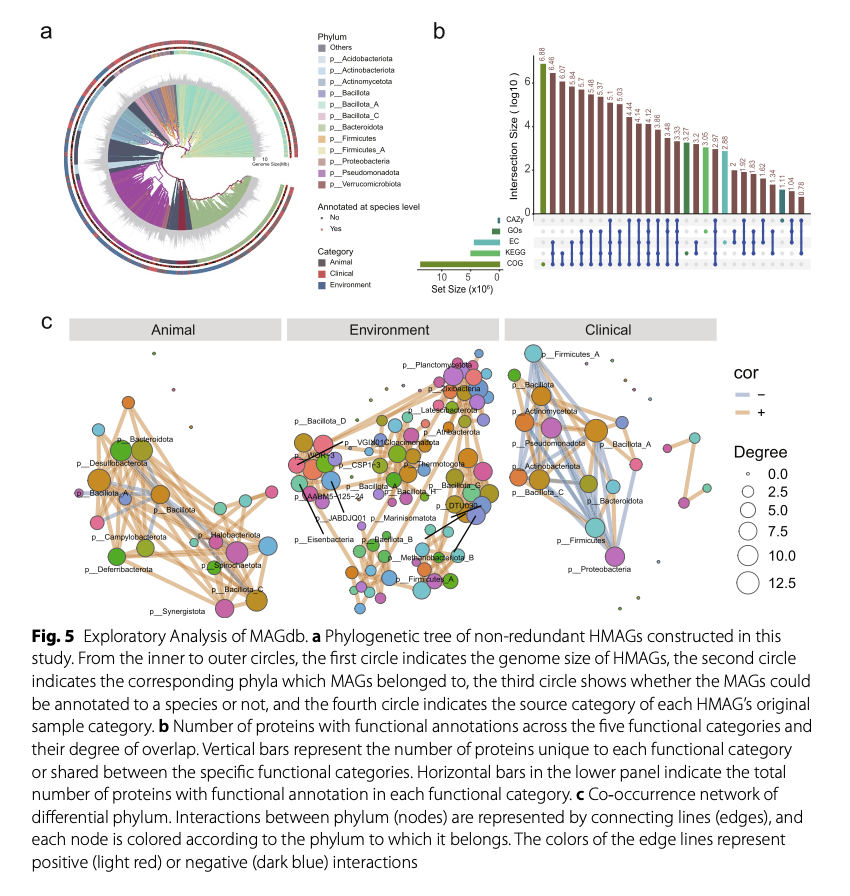
Functional annotation analysis indicates that 94% of the genes in MAGdb can be annotated to at least one mainstream functional database (such as COGs, KEGG, GO, CAZy, etc.), revealing the significant functional potential of these microorganisms in metabolism, signal transduction, and carbohydrate utilization (Figure 5b). Additionally, the team constructed co-occurrence networks of microbial phyla under different environments. The analysis found that in the animal gut, different microbial phyla mainly exhibit positive correlations (co-occurrence), while in clinical samples (such as the human gut), they mainly show negative correlations (competitive exclusion), which may reflect differences in the driving forces of microbial community assembly in different ecological niches (Figure 5c). These analyses not only demonstrate the data quality of MAGdb but also provide new scientific hypotheses for future research.
Full Summary and Outlook
MAGdb is a comprehensive, high-quality, and standardized metagenome-assembled genome database that provides a valuable “one-stop” resource for microbiology and metagenomics research by integrating and reanalyzing vast amounts of public data. The establishment of this database greatly facilitates researchers, allowing them to quickly access high-quality microbial genomes from different habitats, thereby bypassing complex and time-consuming data processing steps and focusing on subsequent biological questions. The standardized annotations and detailed metadata of MAGdb enhance the reproducibility and comparability of research, providing a solid foundation for exploring microbial diversity, discovering new species, elucidating microbial functions, and understanding ecological interactions within microbial communities.
The research team stated that MAGdb will continue to be updated and improved. Future plans include incorporating more new metagenomic data, especially from third-generation sequencing (long-read) technologies, which will help obtain more complete and accurate MAGs. Additionally, the database will expand to include viral metagenomes, incorporating viral operational taxonomic units (vOTUs), and developing more online analysis modules to support deeper functional and evolutionary relationship exploration. We have reason to believe that MAGdb will have a positive and far-reaching impact on advancing research in various fields such as microbial ecology, environmental science, and human health.
Research Team and Funding
The first author of the paper is Guo Ye from Zhejiang University School of Medicine. The corresponding authors are researcher Ting-Ting Li from Nanhu Laboratory and professor Tao Li from Zhejiang University School of Medicine. This research was supported by the National Natural Science Foundation, Nanhu Laboratory, and the Central Guidance for Local Science and Technology Development Fund Project.
DOI Link
DOI: 10.1186/s13059-025-03711-6