Source: Yunqi IVD
Original Title: Comprehensive Analysis of Molecular Diagnosis Technology (Part 1, Part 2, Part 3)
Part 1
Molecular diagnosis technology uses molecular biology methods to detect the expression and structure of genetic material from the human body and various pathogens, thereby achieving the purpose of predicting and diagnosing diseases.
In recent years, with the upgrading and iteration of molecular diagnosis technology, the clinical application of molecular diagnosis has become increasingly widespread and in-depth, entering a period of rapid development in the molecular diagnosis market.
The author summarizes the common molecular diagnosis technologies on the market, divided into three parts: the first part introduces PCR technology, the second part introduces nucleic acid isothermal amplification technology, and the third part introduces sequencing technology.
PCR Technology
PCR (Polymerase Chain Reaction) is one of the in vitro DNA amplification technologies, with a history of over 30 years.
PCR technology was first invented in 1983 by Kary Mullis of Cetus Corporation in the United States. In 1985, Mullis applied for the PCR patent, and in the same year published the first academic paper on PCR in Science. In 1993, Mullis received the Nobel Prize in Chemistry for this.
Basic Principle of PCR
PCR can amplify target DNA fragments by more than a million times. The principle is that under the catalysis of DNA polymerase, using the mother strand DNA as a template, and specific primers as the starting point for extension, it replicates complementary daughter strand DNA to the mother strand template DNA through denaturation, annealing, and extension steps.

The standard PCR process consists of three steps:
1. Denaturation: Using high temperature to separate the DNA double strands. The hydrogen bonds between the DNA double strands are broken at high temperatures (93-98°C).
2. Annealing: After the DNA double strands are separated, the temperature is lowered to allow the primers to bind to the single-stranded DNA.
3. Extension: DNA polymerase starts synthesizing the complementary strand from the primer that binds during cooling, completing a cycle and doubling the number of DNA fragments.
Repeating these three steps 25-35 times will result in an exponential increase in the number of DNA fragments.
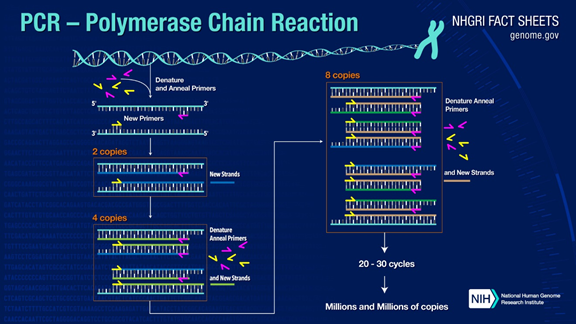
The cleverness of PCR lies in designing different primers for different target genes, allowing the target gene fragments to be amplified to the millions level in a short time.
Currently, PCR can be divided into three categories: conventional PCR, quantitative fluorescence PCR, and digital PCR.
First Generation Conventional PCR
Conventional PCR amplification uses conventional PCR amplifiers to amplify target genes, and then uses agarose gel electrophoresis to detect the products, which can only perform qualitative analysis.
Major drawbacks of the first-generation PCR:
-
Prone to non-specific amplification and false-positive results.
-
Detection takes a long time and the operation is cumbersome.
-
Can only perform qualitative detection.
Second Generation Quantitative Fluorescence PCR
Quantitative fluorescence PCR (Real-Time PCR), also known as qPCR, monitors the accumulation of amplification products through the accumulation of fluorescence signals by adding fluorescent probes that indicate the progress in the reaction system. The results can be judged through the fluorescence curve, and quantitative detection can be performed using the Cq value and standard curve.
The qPCR technology, due to its operation in a closed system, reduces the probability of contamination and allows for quantitative detection through monitoring fluorescence signals, making it the most widely used technology in clinical applications and the dominant technology in PCR.
The fluorescent substances used in real-time quantitative PCR can be divided into: TaqMan fluorescent probes, molecular beacons, and fluorescent dyes.
1) TaqMan Fluorescent Probes:
During PCR amplification, a specific fluorescent probe is added simultaneously with a pair of primers. This probe is an oligonucleotide, with a reporter fluorescent group and a quencher fluorescent group labeled at both ends.
When the probe is intact, the fluorescence signal emitted by the reporter group is absorbed by the quencher group; during PCR amplification, the 5′-3′ exonuclease activity of Taq enzyme degrades the probe, separating the reporter fluorescent group from the quencher fluorescent group, allowing the fluorescence monitoring system to receive the fluorescence signal. Thus, for each amplified DNA strand, a fluorescent molecule is formed, achieving synchronization of fluorescence signal accumulation with PCR product formation.
2) SYBR Fluorescent Dyes:
Excess SYBR fluorescent dye is added to the PCR reaction system. SYBR fluorescent dye non-specifically incorporates into double-stranded DNA and emits a fluorescence signal, while SYBR dye molecules that do not incorporate into the strands do not emit any fluorescence signal, ensuring that the increase in fluorescence signal is completely synchronized with the increase in PCR products. SYBR binds only to double-stranded DNA, so specificity can be determined by the melting curve.
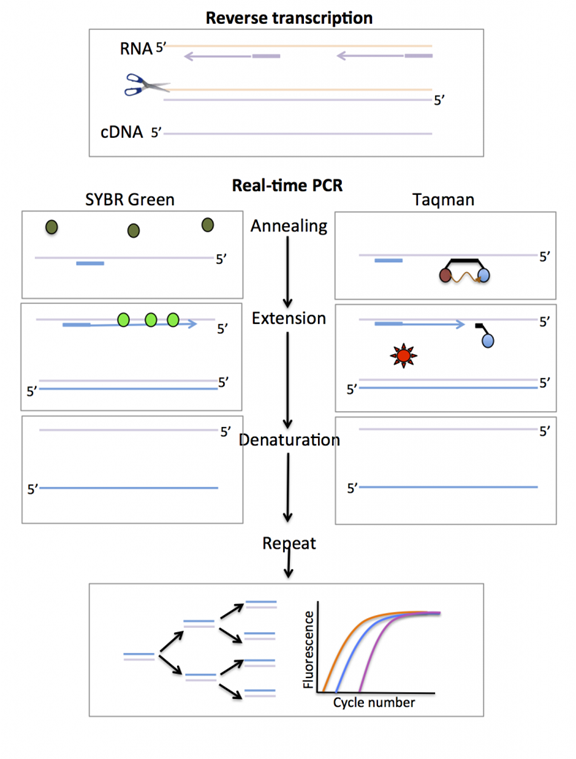
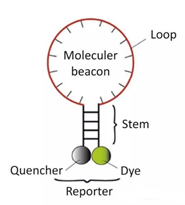
3) Molecular Beacons:
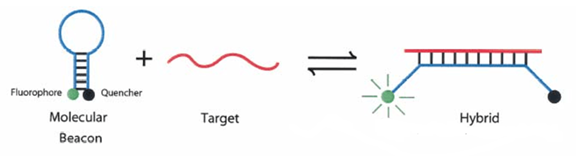
A molecular beacon is a dual-labeled oligonucleotide probe that forms a hairpin structure of about 8 bases at both ends. The nucleic acid sequences at both ends are complementary, bringing the fluorescent group and the quencher group close together, preventing fluorescence.
After the PCR products are generated, during the annealing process, the middle part of the molecular beacon pairs with a specific DNA sequence, separating the fluorescent gene from the quencher gene, producing fluorescence.
Major drawbacks of the second-generation PCR:
-
Sensitivity is still lacking; low copy specimens are not accurately detected.
-
Background values can interfere, easily affecting results.
-
Detection results can be easily disturbed when PCR inhibitors are present in the reaction system.
Third Generation Digital PCR
Digital PCR (dPCR) calculates the copy number of target sequences through endpoint detection, allowing for precise absolute quantitative detection without the need for internal references or standard curves.
Digital PCR uses endpoint detection, which is not dependent on Ct values (cycle threshold), thus reducing the impact of amplification efficiency on the reaction and improving tolerance to PCR inhibitors, achieving high accuracy and reproducibility.
Due to its high sensitivity and accuracy, and its resistance to interference from PCR reaction inhibitors, it can achieve true absolute quantification without standard products, making it a hot topic in research and application.
Based on the different forms of the reaction units, it can be divided into three main types: microfluidic, chip-based, and droplet-based systems.
1)Microfluidic Digital PCR, mdPCR.
Based on microfluidic technology, it segments the DNA template. Microfluidic technology can achieve the generation of samples at the nanoscale or smaller droplets, but the droplets need special adsorption methods to combine with the PCR reaction system. mdPCR has gradually been replaced by other methods.
2)Droplet Digital PCR, ddPCR.
This method uses oil-in-water droplet generation technology to process samples into droplets, dividing the reaction system containing nucleic acid molecules into thousands of nanoscale droplets, each of which either does not contain the target nucleic acid molecule or contains one to several target nucleic acid molecules.
Taking Bio-Rad’s ddPCR as an example, it mainly includes three steps:

First, prepare samples and generate droplets. As shown in the figure, an 8×3 microplate, one row adds samples, and one row adds oil droplets, generating 20,000 droplets per sample through the droplet generator.

Second, perform “oil-in-water” PCR, placing the microplate into the PCR amplifier to perform 40 thermal cycles of PCR reaction for each droplet.
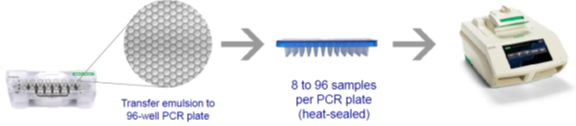
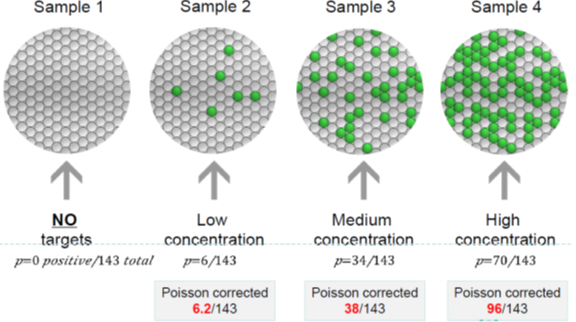
Third, read droplet results, placing the microplate into the reading instrument to obtain fluorescence signals of each droplet’s PCR endpoint results through flow cytometry technology, and correct the results using Poisson distribution.
The Bio-Rad QX ONE digital PCR system launched last year integrates droplet generation, thermal cycling reactions, and droplet reading steps, minimizing manual operations.
Equipped with four independent fluorescence detection channels, combined with ddPCR’s unique high-order multiplex PCR technology, it can achieve 8-fold copy number variation (CNV) detection, 5-fold mutation detection, or 4-fold gene expression detection in a single reaction well.

3)Chip-based Digital PCR, cdPCR.
Using integrated fluidic pathways technology, many microtubes and microchambers are etched on silicon or quartz glass, controlling the flow of the solution through different control valves, dividing the sample liquid into uniformly sized nanoscale reaction wells for digital PCR reactions, achieving absolute quantification.
Taking the cdPCR technology from France’s Stilla Technologies as an example, it mainly includes the following steps:
First, sample addition and droplet generation: Add samples and PCR reaction liquid to the microfluidic chip, place it in the instrument, which can generate a single-layer array of 20,000-30,000 droplets through physical methods.
Second, perform PCR amplification for each droplet: PCR amplification is automatically performed in the Naica Geode droplet generation amplification system.
Third, read and analyze results: Place the chip on the Prism droplet reading analysis system for fluorescence signal collection, counting positive and negative droplets, and calculating the absolute copy number concentration of the target gene using Poisson distribution.
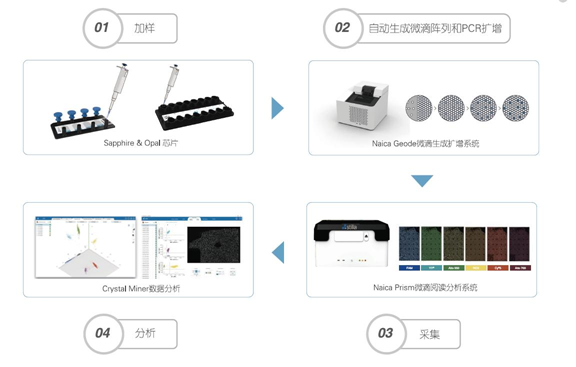
To summarize, add samples and PCR reaction liquids to the microfluidic chip, place it in the instrument, generate a single-layer array of droplets through physical methods, then amplify each droplet, followed by six-channel fluorescence signal collection, counting positive and negative droplets, and calculating the absolute copy number concentration of the target gene using Poisson distribution.
Major drawbacks of the third-generation PCR:
-
Instruments and reagents are expensive.
-
Template quality requirements are high; an excess of template volume will lead to an inability to quantify, while too little will reduce quantification accuracy.
-
Non-specific amplification can also produce false positives.
PCR’s Various Extension Technologies
1. Decremental PCR (touchdown PCR): The temperature gradually decreases in the first few cycles.
2. Reverse Transcription PCR (RT-PCR): Uses cDNA reverse transcribed from mRNA as a template, and since it is amplified from phenotypic genes, the resulting cDNA product does not carry introns (segments in genes without meaning), commonly used in molecular cloning technology.
3. Hot Start PCR: Uses heat-activated DNA polymerase to reduce non-specific products.
4. Nested PCR: First amplifies several cycles with low-specificity primers to increase template quantity, then amplifies with high-specificity primers.
5. Multiplex PCR: Uses multiple sets of primers in the same tube.
6. Reconditioning PCR: Dilutes PCR products 10 times, then reintroduces original concentration primers and dNTPs to cycle three times to eliminate heterodimers in the products.
7. dsRNA Synthesis (dsRNA replicator): Combines high-fidelity DNA polymerases, T7 RNA polymerase, and Phi6 RNA replicase; transcribes from double-stranded DNA to corresponding double-stranded RNA (dsRNA). Can be applied in RNAi experimental operations.
8. COLD-PCR (co-amplification at lower denaturation temperature PCR): A PCR application technique used to detect mutations or specific alleles.
Part 2
Continuing from the first part, this part introduces isothermal amplification technology.
PCR is the most widely used nucleic acid amplification technology, with its sensitivity and specificity widely applied. However, PCR requires repeated thermal denaturation, limiting its application in clinical field detection due to reliance on instrument equipment.
Since the early 1990s, many laboratories have begun to develop isothermal amplification technologies that do not require thermal denaturation, and various techniques such as loop-mediated isothermal amplification (LAMP), strand displacement amplification (SDA), rolling circle amplification (RCA), and nucleic acid sequence-based amplification (NASBA) have been developed.
Loop-Mediated Isothermal Amplification
Loop-mediated isothermal amplification (LAMP) is a new nucleic acid amplification technology first proposed by Notomi et al. from Eiken Chemical Company in Japan in 2000.
Based on this technology, Eiken also had patent disputes with a domestic company.
The amplification principle is based on DNA being in a dynamic equilibrium state at around 65°C. When any primer pairs with the complementary site of double-stranded DNA for base pairing extension, the other strand will dissociate, becoming single-stranded.
At this temperature, using four specific primers and a strand-displacing DNA polymerase, the synthesis of strand-displaced DNA is continuously self-cycling.
First, determine six specific regions F3, F2, F1, B1, B2, B3 on the target gene, then design four primers based on these six specific regions (as shown in the figure):
The forward inner primer (FIP) is composed of F1c and F2. The backward inner primer (BIP) is composed of B1c and B2, with TTTT as a spacer in between.
The outer primers F3 and B3 are composed of the F3 and B3 regions on the target gene.
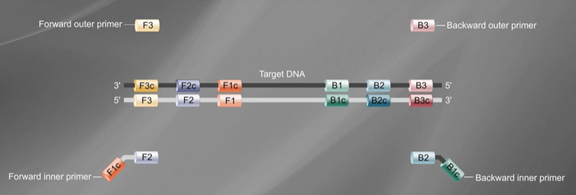
In the LAMP reaction system, the concentration of inner primers is several times higher than that of outer primers. The inner primers first bind to the template strand to synthesize the complementary strand, forming double-stranded DNA. Subsequently, the outer primers bind to the template strand to form double-stranded DNA, under the action of Bst DNA polymerase, releasing the complementary strand synthesized by the inner primers. This complementary strand undergoes a series of reactions to ultimately form a DNA single strand with a dumbbell structure.
Using the dumbbell-structured DNA single strand itself as a template, a transitional stem-loop structure DNA with one end open is continuously formed, guided by the inner and outer primers, leading to continuous strand-displacing extension reactions, ultimately forming a mixture of DNA with multiple stem-loop structures of varying lengths.
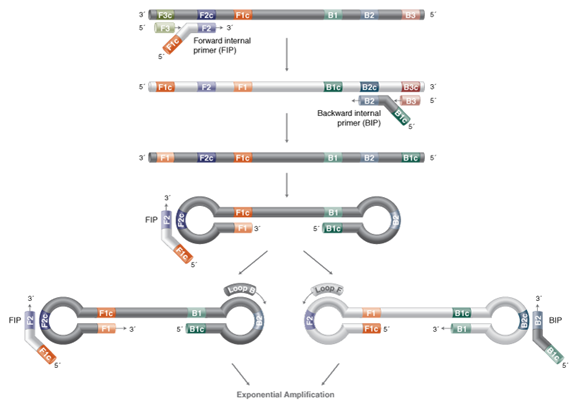
The advantages and disadvantages of loop-mediated isothermal amplification (LAMP):
The advantages of LAMP:
(1) High amplification efficiency, capable of effectively amplifying 1-10 copies of the target gene within 1 hour, with an amplification efficiency 10-100 times that of conventional PCR.
(2) Short reaction time, high specificity, and does not require special equipment.
The disadvantages of LAMP:
(1) High requirements for primers.
(2) Amplification products cannot be used for cloning and sequencing, only for judgment.
(3) Due to its high sensitivity, it is prone to aerosol formation, leading to false positives that affect detection results.
Strand Displacement Amplification
Strand displacement amplification (SDA) is an enzyme-based in vitro isothermal amplification technology for DNA proposed by American scholar Walker in 1992.
The basic system of SDA includes a restriction endonuclease, a DNA polymerase with strand displacement activity, two pairs of primers, dNTPs, and calcium, magnesium ions, and a buffer system.
The principle of SDA is based on the target DNA having chemically modified restriction endonuclease recognition sequences at both ends. The endonuclease opens a gap at its recognition site, and DNA polymerase extends the gap at the 3′ end and replaces the next DNA strand.
The replaced single-stranded DNA can bind with the primers and be extended by DNA polymerase to form double-stranded DNA. This process continues to repeat, efficiently amplifying the target sequence.
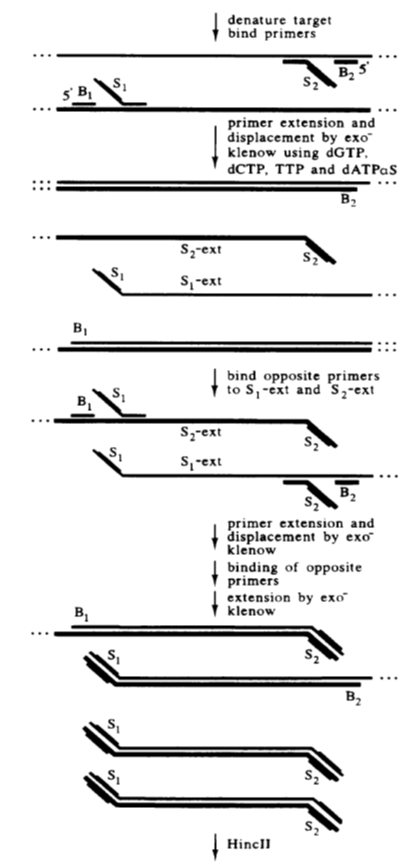
The advantages and disadvantages of strand displacement amplification (SDA):
The advantages of SDA:
High amplification efficiency, short reaction time, high specificity, and does not require special equipment.
The disadvantages of SDA:
Inhomogeneous products; during SDA cycles, some single and double-stranded products are always generated, leading to tailing phenomena when detected by electrophoresis.
Rolling Circle Nucleic Acid Amplification
Rolling circle amplification (RCA) is proposed by referencing the rolling circle replication of pathogenic organisms’ DNA, referring to the amplification of target genes at constant temperature using single-stranded circular DNA as a template, under the action of special DNA polymerases (such as Phi29).
RCA can be divided into linear amplification and exponential amplification, with linear RCA efficiency reaching 105 times, while exponential RCA can reach 109 times.
To distinguish simply, as shown in the figure, linear amplification a uses only one primer, while exponential amplification b uses two primers.
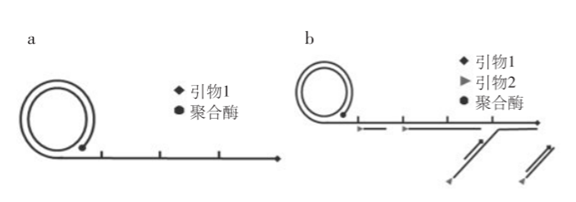
Linear RCA, also known as single-primer RCA, binds one primer to circular DNA, extended under the action of DNA polymerase, producing a linear single strand of massive repetitive sequences thousands of times long.
Since the product of linear RCA is always connected to the starting primer, its signal is easily fixed, which is a significant advantage.
Exponential RCA, also known as hyperbranched RCA (HRCA), uses one primer to amplify RCA products; the second primer hybridizes with the RCA product and extends, displacing the downstream primer chain already bound to the RCA product, repeatedly extending and displacing, producing branched RCA amplification products.
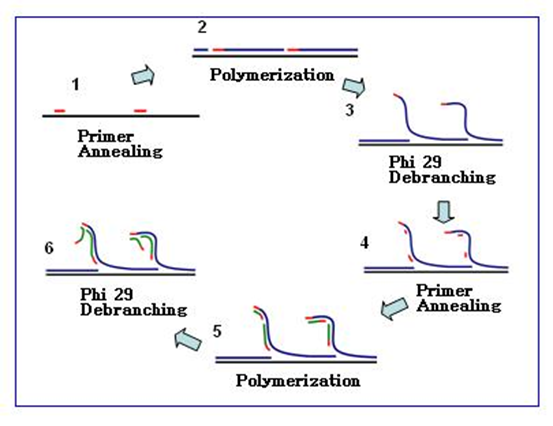
For example, the TruePrime rolling circle amplification kit from 4BaseBio uses TthPrimPol and Phi29 DNA polymerase to achieve amplification without adding primers.
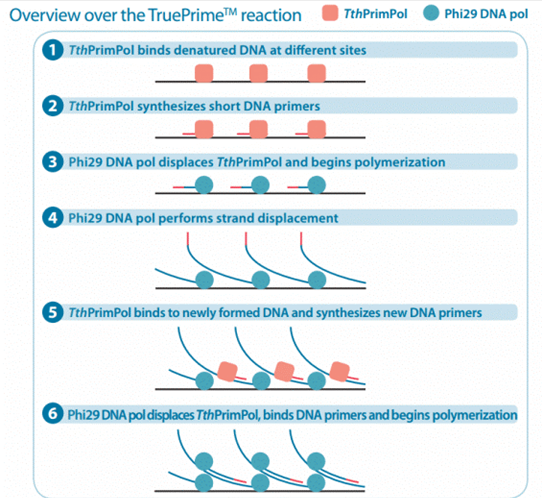
The advantages and disadvantages of rolling circle amplification (RCA):
The advantages of RCA:
High sensitivity, good specificity, and easy operation.
The disadvantages of RCA:
Background issues during signal detection. During the RCA reaction, unlooped lock probes and unbound probes’ template DNA or RNA may produce some background signals.
Nucleic Acid Sequence-Based Amplification Technology
Nucleic acid sequence-based amplification technology (NASBA) is a new technology developed on the basis of PCR, guided by a pair of primers with T7 promoter sequences, enabling continuous, isothermal nucleic acid amplification that can amplify template RNA by about 109 times in approximately 2 hours, which is 1000 times higher than conventional PCR methods, and does not require special instruments.
This technology was quickly used for rapid disease diagnosis as soon as it appeared, and many companies’ RNA detection kits use this method.
Although RNA amplification can also use reverse transcription PCR technology, NASBA has its advantages: it can be performed under relatively constant temperature conditions and is more stable and accurate than traditional PCR technology.
The reaction takes place at 41 degrees Celsius and requires AMV (avian myeloblastosis virus) reverse transcriptase, RNase H, T7 RNA polymerase, and a pair of primers to complete.
The process mainly includes:
-
The forward primer contains a T7 promoter complementary sequence, during the reaction, the forward primer binds to the RNA strand, catalyzed by AMV enzyme to form DNA-RNA double strands.
-
RNase H digests the RNA in the hybrid double strand, retaining the DNA single strand.
-
Under the action of the reverse primer and AMV enzyme, a DNA double strand containing T7 promoter sequences is formed.
-
Under the action of T7 RNA polymerase, the transcription process is completed, producing a large amount of target RNA.
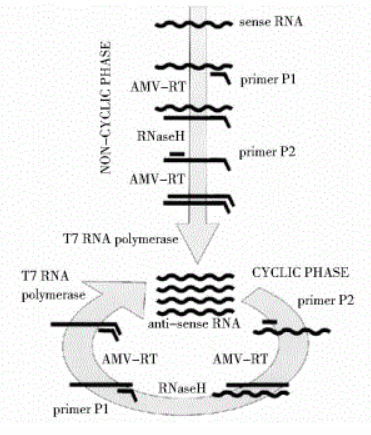
The advantages of NASBA:
(1) Its primers carry T7 promoter sequences, while foreign double-stranded DNA lacks T7 promoter sequences, making amplification impossible; thus, this technology has high specificity and sensitivity.
(2) NASBA directly merges the reverse transcription process into the amplification reaction, shortening the reaction time.
The disadvantages of NASBA:
(1) The reaction components are relatively complex.
(2) Requires three enzymes, resulting in higher reaction costs.
Part 3
This part, as the final part of the comprehensive analysis of molecular diagnosis technology, introduces sequencing technology.
Gene sequencing technology began in 1977, when Sanger invented the DNA double-deoxy termination sequencing method, marking the beginning. Sanger won the Nobel Prize in Chemistry twice, in 1958 and 1980, for insulin sequencing and DNA sequencing, respectively, making him the fourth person to receive the Nobel Prize twice and the only person to receive the Nobel Prize in Chemistry twice.

Sanger Sequencing
Sanger sequencing uses four double-deoxy ribonucleic acids (ddNTPs) added to the growing chain, and because double-deoxy ribonucleic acid lacks one oxygen atom, once added to the DNA chain, the reaction terminates.
By constructing four reaction systems and adding AGCT four types of double-deoxy ribonucleic acids, while adjusting the relative concentrations of deoxyribonucleic acid (dNTP) and ddNTP, products terminating the reaction can be amplified to hundreds or thousands of bases.
Subsequently, the products are separated by gel electrophoresis, divided into four lanes, each corresponding to one base, and then the banding results are read.
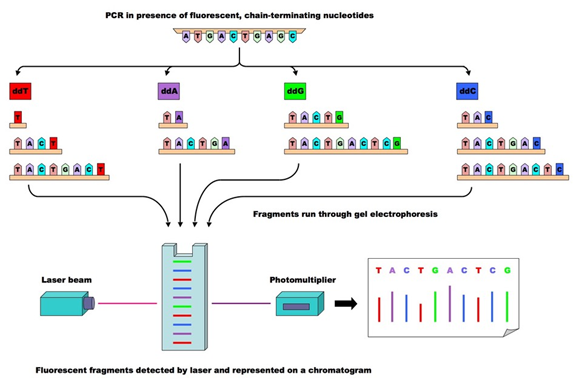
This method is known as the gold standard for gene detection due to its high accuracy, but it is time-consuming and costly.
The Human Genome Project in 2001 used Sanger sequencing to complete it. Since the establishment of the Human Genome Project in 1990, scientists worldwide spent 11 years and $3 billion to finish it.
Entering the 21st century, with the development of physical and chemical technologies, fluorescent groups with the same excitation wavelength but different emission wavelengths began to be used to label ddNTPs, producing different colors of light corresponding to ATGC, which are read by computers, greatly improving sequencing speed and efficiency.
Second Generation Sequencing
Second-generation sequencing technology, also known as high-throughput sequencing (HTS) technology, enables large-scale parallel sequencing compared to first-generation sequencing. The basic principle is to divide the genome into short fragments, sequence the short fragments, and then splice them together.
Compared to first-generation sequencing technology, it has advantages such as high throughput and low cost. Currently, the cost for the same amount of data detection is about 0.01% of first-generation sequencing technology, greatly promoting the application of sequencing technology in clinical detection.
In 2005, 454 Company launched the Genome Sequencer 20 System (GS 20) based on pyrosequencing, marking the beginning of high-throughput sequencing. In 2007, Roche acquired 454 and launched a series of more advanced NGS systems, greatly enhancing sequencing throughput and accuracy.
Despite the advantage of read length, sequencing throughput and cost have always limited the promotion of the 454 platform, with the cost for the same amount of data being about 100 times that of Illumina, leading Roche to terminate 454 NGS sequencing-related businesses at the end of 2016.
In 2006, Solexa Company launched the Genome Analyzer system, incorporating DNA clusters, bridge PCR, and reversible terminator technologies, giving the GA system significant advantages in high throughput, low cost, and wide application range. In 2007, Illumina acquired Solexa and released second-generation sequencers.
Second-generation sequencing has matured over the years, and currently, the market can be divided into four types based on sequencing technology: sequencing by synthesis (Illumina), semiconductor sequencing (ThermoFisher), combined probe anchored polymerase sequencing (BGI), and pyrosequencing.
Illumina Sequencing by Synthesis
The Illumina sequencing process mainly includes sample preparation, cluster generation, sequencing, and data analysis.
First, the nucleic acid is extracted using DNA or RNA extraction kits, and then it is randomly fragmented into lengths of about 90-250 bp or controlled to keep all DNA within a certain length range.
Specific sequences are added to these DNA fragments for subsequent amplification and sequencing. As shown in the figure, the areas complementary to the flow cell primers (P5, P7), the regions binding to Read 1 and Read 2 sequencing primers (Rd1SP, Rd2 SP), and the index sequence area are shown.
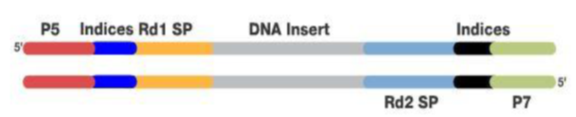
The DNA collection with added adapter sequences is called the DNA library, completing the library preparation, which can be done using commercial library preparation kits.
The second step is cluster generation.
Cluster generation is the process in which the above DNA fragments are amplified, completed in the flow cell, which is a thick glass slide containing eight channels, each randomly implanted with short DNA fragments that can bind to the library adapters P5 or P7.
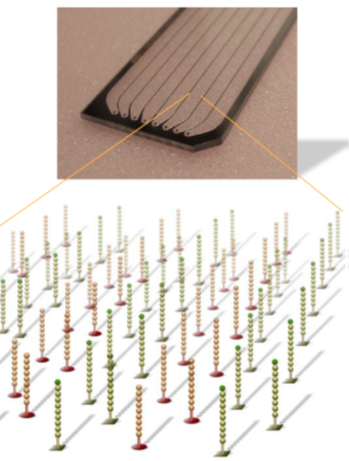
First, the primers and fixed DNA fragments in the flow cell complementarily pair and fix to the channel surface. Then, under the action of DNA polymerase, the DNA chain undergoes complementary extension to form double-stranded DNA. Through denaturation, single strands are eluted, leaving one single strand that links to adjacent fixed adapters, forming a single-stranded bridge.
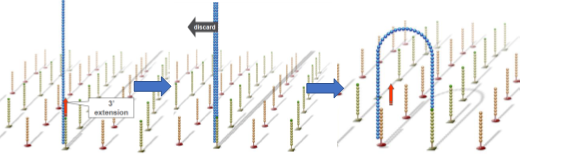
Similarly, the single-stranded bridge is extended and paired under the action of DNA polymerase to form a double-stranded bridge. Through denaturation, two single strands are formed, and these two single strands each bind to adjacent fixed primers, forming two single-stranded bridges. This cycle repeats, ultimately forming millions of DNA clusters.
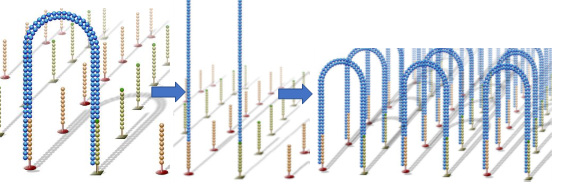
All DNA fragments undergo amplification, and after amplification, the reverse link is cut and eluted, leaving only the forward strand, while the 3′ end is blocked to prevent reformation of single-stranded bridges through complementary binding.
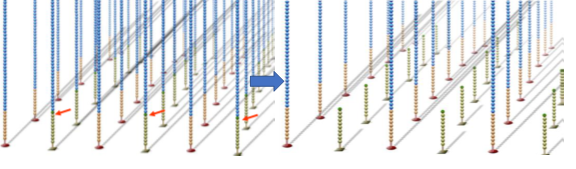
The third step is sequencing.
First, fluorescently labeled dNTPs and enzymes are added to the flow cell, starting synthesis from the primers to produce complementary strands. Due to the presence of azide groups at the 3′ end of dNTPs, only one base can be measured per cycle. After one base is synthesized, excess dNTPs and enzymes are washed away, and laser scanning is used to obtain fluorescence signals.
Subsequently, reagents are added to remove the azide groups and fluorescent groups, and then fluorescently labeled dNTPs and enzymes are introduced again, starting synthesis of another base from the primers. This process is repeated to complete the first read.
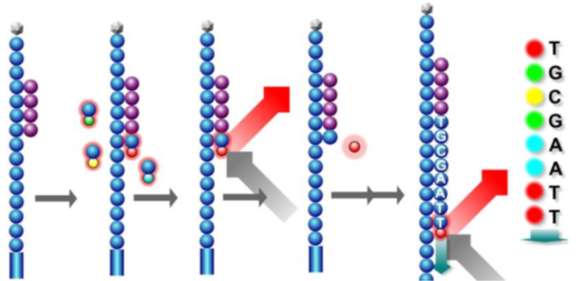
All DNA fragments have one base read simultaneously, and during large-scale parallel processes, the images read by machines are similar to the one below:

At the same time, different indexes are added to distinguish each sample and positive/negative strands. After the first read is completed, the copied strands are washed away, and index fragment primers are introduced to hybridize with templates. After completing the sequence reading, they are washed away. The sequences read can then be labeled by comparing with the known indexes at the start, facilitating subsequent analysis.
Paired-end sequencing is now mainstream; to complete double-end sequencing, the template strand must first be deprotected at the 3′ end, fold the template, introduce index fragments, and form double-stranded bridges with the participation of polymerase. Then denature to restore to single strands, remove the forward strand, and wash it away, leaving the reverse strand, which undergoes similar processes to complete the reading.
The fourth step is data analysis.
After sequencing, millions of reads are generated, and sequences from different samples are classified based on the indexes constructed during sample preparation. For each sample, bases with similar extensions are clustered together. The forward and reverse reads are paired to generate contiguous sequences. These sequences are matched against a reference genome to achieve complete sequence construction.
Thermo Fisher Semiconductor Sequencing
The Ion Torrent platform from Thermo Fisher is a high-throughput sequencer based on semiconductor technology. This platform uses a high-density semiconductor chip filled with small holes, each small hole serving as a sequencing reaction pool, with sensors at the bottom.

The core technology of sequencing is to read information using semiconductor technology; during sequencing, whenever a base binds, H+ is released, causing a change in potential that can be detected. By detecting hydrogen ions and converting them into electrical signals, real-time base reading is achieved.
The sequencing process mainly includes library construction, oil-in-water PCR, sequencing, and data analysis.
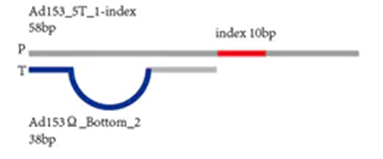
First, during library construction, unlike Illumina’s adapters with protruding T bases at the 3′ end, flat-end adapters (P1) and X or A adapters are added to both ends of the DNA. The X adapter carries an index, while the A adapter does not.
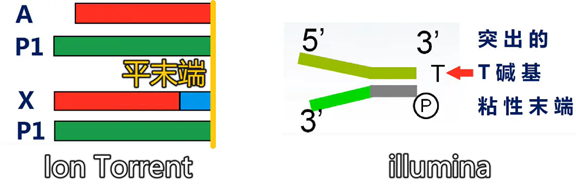
The X adapter carries a barcode sequence (the blue sequence near the end), while the A adapter does not carry a barcode sequence.
The advantage of the X adapter is that it can allocate the sequencing throughput of one chip to multiple libraries, and after sequencing, it can be distinguished by the barcode.
The advantage of the A adapter is that it directly measures the sample sequence, which is undoubtedly better for fully utilizing the sequencing read length. However, its disadvantage is that it does not have a barcode, so one chip can only hold one sample.
AmpliSeq is the library construction scheme on the Ion Torrent platform, which is primarily based on multiplex PCR methods to amplify multiple DNA fragments to be sequenced from samples at once, then convert them into libraries for sequencing.
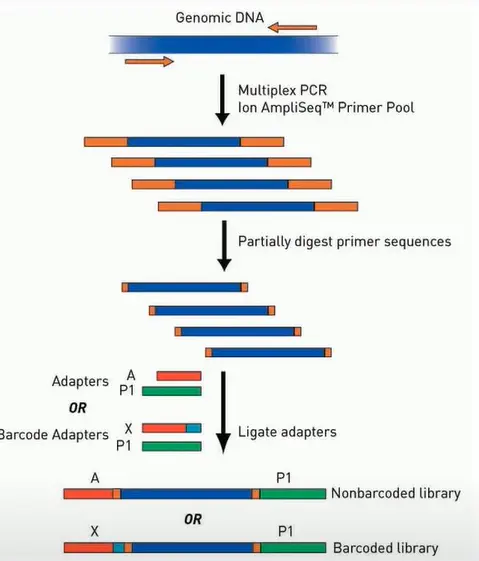
The ends of the PCR products are 20-30 bp bases that are the sequences of PCR primers. If these are sequenced, a significant portion of the sequencing read length and data will be wasted. Therefore, a special chemical modification is designed on this primer, which can be digested by Fupa reagents, allowing for as much sample sequence to be sequenced as possible.
Emulsion PCR or oil-in-water PCR
Before sequencing on Ion Torrent, the library needs to be combined with sequencing beads and amplified. This method is called oil-in-water PCR, also known as Emulsion PCR.
The EP tube contains oil and water phases, with the water phase being the core, and the oil phase serving as a separator. The water phase includes the library, primers, enzymes, MasterMix, sequencing beads, and other main components of the PCR reaction.
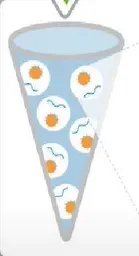
The diameter of sequencing beads is about 1-2.4 microns, and each oil-in-water PCR contains many beads. The surface of these beads covalently connects many PCR primers, complementing the P1 sequence.
At the same time, free PCR primers in the oil-in-water PCR have sequences consistent with A/X adapters, and the 5′ ends are all labeled with biotin.
Mix primers, enzymes, sequencing beads, etc. in the water phase first, then add oil, mixing to form an emulsion, where the oil separates the water phase into small droplets. After PCR reactions, copies of the DNA library will grow on the surface of the beads.
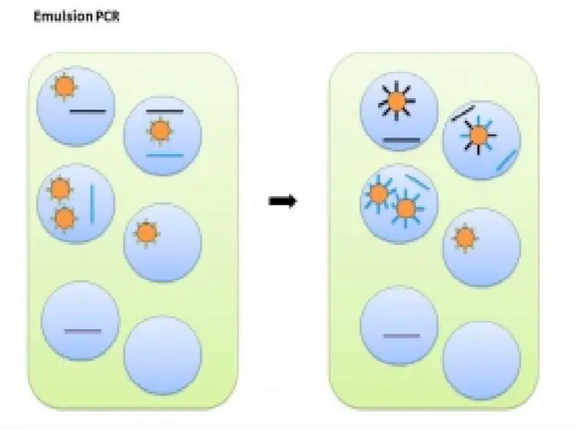
Then, magnetic beads labeled with streptavidin are mixed with the beads. The beads where PCR has occurred will bind to the magnetic beads due to the biotin on their primers, while the beads where PCR has not occurred will not bind. Next, use a magnet to enrich the effective beads, wash away the non-PCR beads in the supernatant, and finally use elution solution to separate the beads from the magnetic beads for sequencing.
The sequencing mainly occurs on a semiconductor chip, which has millions of tiny holes, each of which serves as a container for sequencing beads and at the same time as a miniature pH meter.
Each small hole can accommodate one sequencing bead. When DNA polymerase polymerizes nucleotides onto the extending DNA chain, a hydrogen ion is released, causing a change in pH in the reaction pool. The ion sensor at the bottom of the pool detects the signal, converting the chemical signal directly into a digital signal, thus reading the DNA sequence.

Data analysis
Solutions containing A, C, G, and T dNTPs are sequentially flowed over the surface of the chip.
For example, if a dCTP solution flows in, and there is a G base on the template, a polymerization reaction occurs, producing a voltage change that is recorded. If the solution flowing in does not match the base on the template, no polymerization reaction occurs, resulting in no voltage change and no base being recorded.
If there are two identical bases adjacent to each other, two bases can be polymerized onto the DNA chain at once, and the voltage change will double, recording two new bases in the sequence.
BGI Combined Probe Anchored Polymerase Sequencing Method
On March 18, 2013, BGI fully acquired CG (Complete Genomics) Company, starting the journey of BGI sequencers. After years of research and development, BGI has successively launched the BGISEQ-500, BGISEQ-50, MGISEQ-200, MGISEQ-2000M, and MGISEQ-T7 sequencing systems.
The sequencing platform adopts DNB (DNA Nanoball) sequencing technology, with each DNA having a diameter of about 220-240 nm.
The main steps include library preparation, cPAS sequencing, and data analysis.
Sample preparation and library construction
The MGI library construction uses bubble adapters and corresponding amplification products, with long and short adapters, single-end and double-end indexes.
After single-strand separation and circularization of the library, using single-stranded circular DNA as a template, the DNA polymerase amplifies the single-stranded circular DNA by 2-3 orders of magnitude under its action, and the amplification product is called DNB.
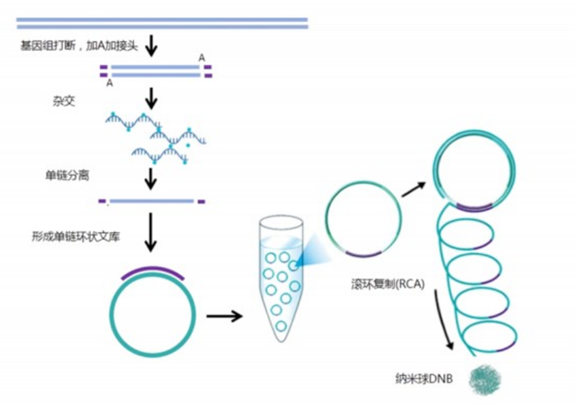
DNB is fixed on an arrayed silicon chip through DNB loading technology to form a nano chip. Since one DNB binds to a small hole on the chip, it repels other DNBs, ensuring that each small hole accommodates only one DNB, preventing signal interference between signal points.
Sequencing
Using semiconductor processing technology, a binding site array is formed on the surface of the modified silicon chip (with a diameter of about 200 nm), achieving the regular arrangement and adsorption of DNA nanoballs. The spacing between arrayed sites is about 700 nm, and each site fixes one DNB, ensuring that light signals between different nanoballs do not interfere with each other.
Read1 primers with fluorescent probes match complementarily on DNBs, and the system collects light signals to obtain the sequence to be sequenced.
MDA (multiple displacement amplification) two-strand sequencing uses random primers to bind to multiple sites with template DNA, initiating replication under the action of Phi29 DNA polymerase, synthesizing DNA along the DNA template while replacing the complementary strand of the template; the replaced complementary strand becomes a new template for subsequent amplification. After the first strand sequencing is completed, the second strand is formed under the action of the enzyme, and second-chain cPAS sequencing is performed.
Third Generation Sequencing Technology
1. PacBio SMRT Single-Molecule Sequencing Technology
Pacific Biosciences was established in 2004 and went public in NASDAQ in 2010, releasing PacBioRS in 2011.
Third-generation gene sequencing technology represented by PacBio sequencing is gradually applied in multiple scientific research fields. This platform is based on single-molecule real-time sequencing technology (SMRT), a single-molecule reading technology based on nanopores that allows for rapid sequence reading without amplification.
Library construction
Because the sequencing read length is long, large fragments (3-10 kb) can be prepared; unlike other second-generation sequencing libraries, this library connects circular single strands at both ends, with single strands connected to double-stranded positive and negative strands, resulting in a structure similar to a dumbbell, called SMRT Bell.
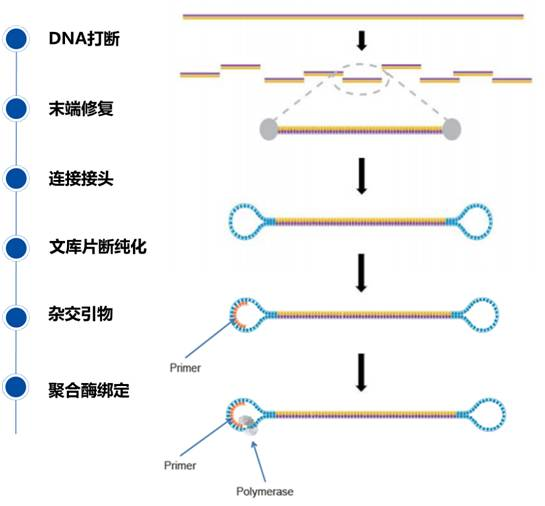
Sequencing
The SMRT Cell chip contains millions of nanoscale zero-mode waveguide pores (ZMWs). A ZMW is a pore with a diameter of only 10-50 nm. After annealing the primers at the single-stranded circular part of the template, this double-stranded part can bind to the polymerase fixed at the bottom of the ZMW.
Each ZMW can contain one DNA polymerase and a DNA sample strand for single-molecule sequencing. The four types of dNTPs carry different fluorescent labels. When a laser hits the bottom of the ZMW, it illuminates a very small area, fixing the DNA polymerase in this area.
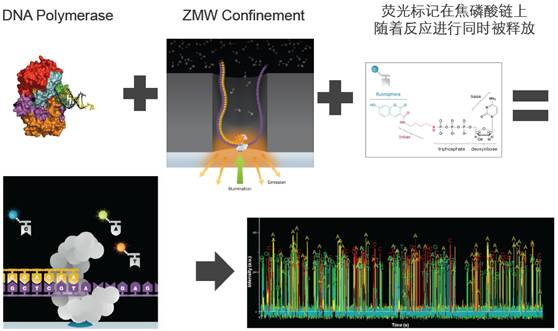
Only in this area can the fluorescent groups carried by bases be activated to be detected. As DNA synthesis proceeds, the next base is extended, and the fluorescent group on the previous dNTP is removed, ensuring continuity in detection.
Different bases emit different lights, and based on the wavelength and peak of the light, the type of base can be determined.
2. Nanopore Sequencing Technology
In 2005, Oxford Nanopore Technologies was established, focusing on the commercial transformation of nanopore sequencing technology. In 2014, the Nano space single-molecule sequencing technology was released.
The nanopore single-molecule sequencing technology developed by Oxford Nanopore is different from previous sequencing technologies; it is based on electrical signals rather than optical signals.
The core of nanopore sequencing technology is a polymer membrane integrating multiple transmembrane channel proteins (i.e., nanopore proteins). By applying voltage across the membrane, a stable current passing through the nanopore is generated. When other objects pass through the nanopore, they affect the current, resulting in recognizable changes in electrical signals.
During sequencing, the double-stranded DNA is unwound into single-stranded DNA under the traction of motor proteins and passes through the nanopore proteins (also called reader proteins). Due to the differences in structure and size of the four bases (A, T, C, G), the current exhibits characteristic ionic current changes, allowing for the reading of base sequences by recognizing these electrical signal changes.

Different models of nanopore sequencers have very consistent principles, with their core being nanopore sequencing chips. Conventional nanopore sequencing chips integrate 512 sequencing channels, with each sequencing channel containing four nanopores. Official data shows that a single chip can continuously sequence for 48 hours, generating 20-30G of data.
The emergence of second-generation sequencing has greatly solved the throughput problem, significantly increasing sequencing speed and accuracy while greatly reducing sequencing costs, but the read length is relatively short. In contrast, the third-generation sequencing, characterized by single-molecule, long reads, low cost, and miniaturization, is achieving another revolution in the sequencing field.
References:
1. Liu Wantong et al., Clinical Application Progress of Molecular Diagnosis Technology
2. Yang Liu et al., Research Progress on Gene Detection Technology for Non-Small Cell Lung Cancer
3. G. Terrance Walker, etc. Strand Displacement Amplification – An Isothermal, In Vitro DNA Amplification Technique
4. Jiang Su, Li Yirong, Principles and Applications of Isothermal Amplification Technology
5. Peng Tao, Nucleic Acid Isothermal Amplification Technology and Its Applications
6. Zhao Luyao et al., Fast Detection Technology Research Based on Loop-Mediated Nucleic Acid Isothermal Amplification
7. England Biolabs, Loop Mediated Isothermal Amplification.
8. G. Terrance Walker, etc. Strand Displacement Amplification Isothermal, In Vitro DNA Amplification Technique.
9. Wu Xiaoliang et al., Research Progress on DNA Rolling Circle Amplification Technology.
10. Li Jinming, “High-Throughput Sequencing Technology”
11. Official websites of various companies
The images above are sourced from the official websites of various companies and public channels; if there is any infringement, please contact us for deletion.