Although the concept of antibody-drug conjugates (ADCs) seems straightforward, developing successful and effective drugs involves numerous challenges. For a successful ADC product, scientists must optimize various parameters, including ADC stability (for instance, the drug payload should not be released before the ADC reaches its target in the body), pharmacokinetics (e.g., the payload should not accumulate in non-target organs), maintaining immunogenicity, and the efficacy of releasing active drugs in the targeted therapeutic area, among others.
Although the concept of antibody-drug conjugates (ADCs) seems straightforward, developing successful and effective drugs involves numerous challenges. For a successful ADC product, scientists must optimize various parameters, including ADC stability (for instance, the drug payload should not be released before the ADC reaches its target in the body), pharmacokinetics (e.g., the payload should not accumulate in non-target organs), maintaining immunogenicity, and the efficacy of releasing active drugs in the targeted therapeutic area, among others.
To enhance tumor specificity, early ADCs predominantly used clinically approved chemotherapeutic drugs as payloads. These chemotherapeutic drugs have undergone comprehensive studies regarding their mechanisms of action and toxicity profiles, including antimetabolites (e.g., methotrexate, 5-fluorouracil), DNA cross-linkers (e.g., mitomycin), and microtubule inhibitors (e.g., vinca alkaloids and taxanes). However, the so-called “first-generation ADCs” were not successful due to issues such as lower payload potency, high expression of target antigens on normal cells, and instability of the linker between the payload and the monoclonal antibody (mAb). For example, one of the early ADCs that failed in clinical trials was the doxorubicin conjugate BR96-DOX. In this ADC, the drug is linked to the linker via a hydrazone bond, while at the other end, the maleimide portion is conjugated to the BR96 antibody through a cysteine residue. However, the relatively unstable hydrazone bond is prone to nonspecific cleavage, and the expression of the antigen target was also observed in normal tissues, resulting in significant toxicity of BR96-DOX in clinical trials.
With continuous technological improvements, more effective payloads have been developed, and antigen targets are carefully selected to enhance selectivity. Ultimately, the first ADC was approved by the U.S. Food and Drug Administration (FDA) in 2000 (Mylotarg™, gemtuzumab ozogamicin). Below, we review the relevant cytotoxic drugs used as ADC payloads.
Mitomycin
Mitomycin is a class of natural products containing a nitrogen mustard structure, derived from Streptomyces caespitosus or Streptomyces lavendulae, with mitomycin A, B, and C being the most notable (Figure 1). Mitomycin C exhibits significant antitumor properties and was clinically applied in 1981.
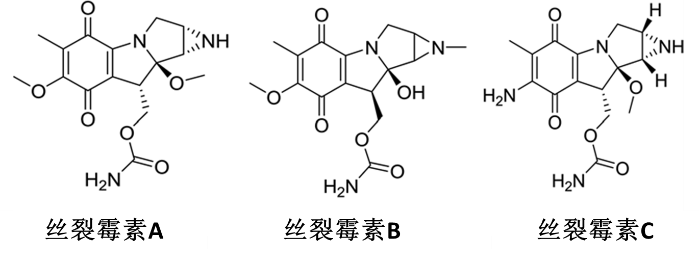
Figure 1 Structure of Mitomycin A, B, and C
So far, many mitomycin analogs have been developed, but mitomycin C itself remains the most well-known and widely studied drug. It can exhibit various biological effects in mammalian cells, such as selective inhibition of DNA synthesis, mutagenesis, causing chromosome damage, and facilitating sister chromatid exchange, all of which make it a potential candidate for cancer treatment. It has been reported that the toxicity of mitomycin C is related to the contents of guanine and cytosine in DNA. Studies show that mitomycin C has strong cytotoxicity against various human tumor cell lines, demonstrating excellent prospects as a chemotherapeutic agent for cancers, including gastric and breast cancer. However, clinical applications of mitomycin C also face numerous challenges, including
(1) Certain cancers (e.g., gastrointestinal cancers) can easily develop resistance to it
(2) Short duration of action
(3) Potential to cause infiltrative ulcers at the injection site
These factors limit the drug’s efficacy in clinical stages.
Mechanism studies indicate that mitomycin C needs to be activated under reducing conditions, after which it reacts with DNA, leading to DNA alkylation and cell killing effects (Figure 2). This reduction-activated characteristic endows mitomycin C with the property of selectively killing specific tumors, especially hypoxic solid tumors. Therefore, early in ADC research, mitomycin C became one of the candidate payloads.
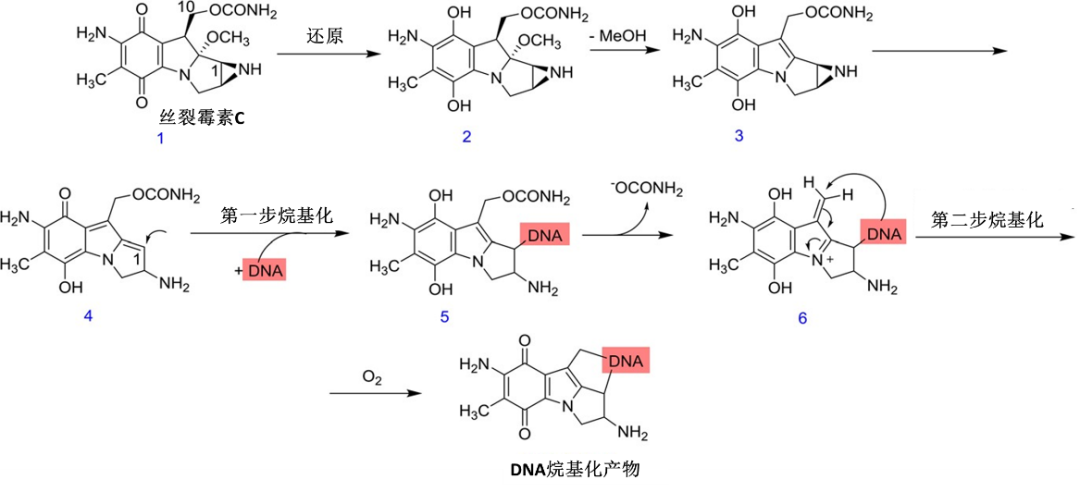
Figure 2 Mechanism of Action of Mitomycin C
However, as of now, there are few examples of mitomycin being used as an ADC payload. One of them is the mitomycin-glucan structure, which is linked to the anti-integrin αvβ3 monoclonal antibody to form the ADC MMCD-mAb. This antibody-drug conjugate was developed by researchers from Kyoto and Nagoya City University in the early 2000s (Figure 3). This ADC demonstrated stronger efficacy and higher safety in treating choroidal neovascularization (CNV) in rat models. In vitro studies of MMCD-mAb showed that after binding to the endothelial cell surface through antigen-antibody interactions, the ADC releases biologically active mitomycin C through chemical hydrolysis. It is also believed that the modified carboxyl glucan, due to its anionic properties, can reduce the uptake of the ADC by the reticuloendothelial system, thereby prolonging the ADC’s circulation half-life. Furthermore, antitumor drugs can be passively targeted to tumor sites by increasing molecular size, utilizing the enhanced permeability and retention (EPR) effect. Therefore, the introduction of a glucan structure with a molecular weight exceeding 70,000 allows MMCD-mAb to cross the vascular wall of CNV and accumulate in the subretinal space near CNV. In summary, data indicate that combining drugs like mitomycin with water-soluble polymers and antibodies targeting neovascularization can enhance efficacy and safety.
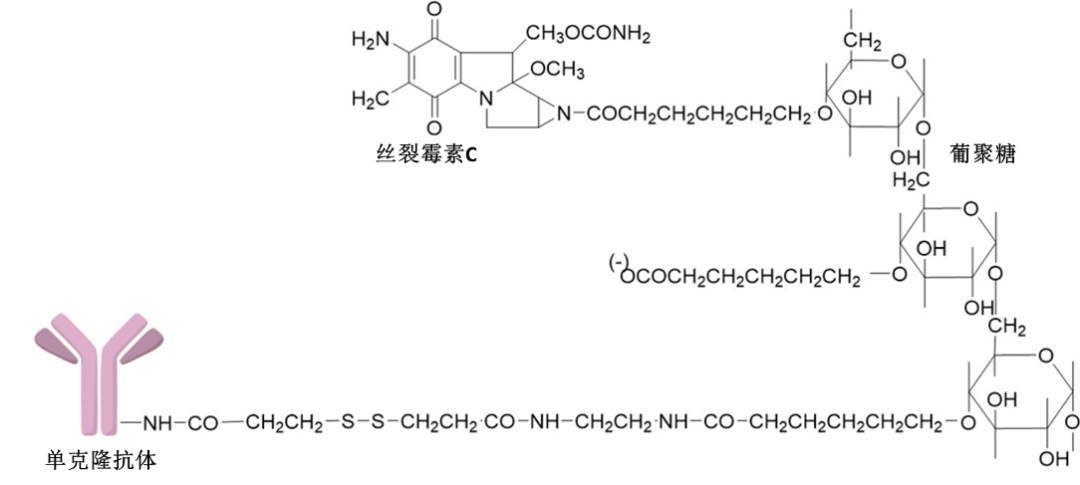
Figure 3 Molecular Structure of ADC MMCD-mAb Containing Mitomycin C (MMC)
The mitomycin molecule is linked to the glucan-containing linker through the nitrogen atom in its nitrogen mustard.
In two other examples of mitomycin-based ADCs, mitomycin C is combined with N-succinyl chitosan and polyethylene glycol chitosan, respectively. These structures have been reported to improve in vitro drug release characteristics and enhance antitumor activity. Although these ADCs have not yet reached the clinical stage, they have already attracted market interest for preclinical and clinical stage research.
Doxorubicin
Doxorubicin is an antitumor drug formed by the combination of an anthraquinone structure and a sugar ring, with its discovery tracing back to the 1950s (Figure 4). At that time, another member of the anthracycline family, daunorubicin, was first isolated from a strain of Streptomyces peucetius. Daunorubicin was very effective in treating acute leukemia and lymphomas; however, it has severe side effects on the body, especially on the heart. Therefore, after genetic modification of the actinobacteria, an improved anthracycline drug, doxorubicin, was generated. Although doxorubicin has a wider therapeutic window than daunorubicin, its cardiac toxicity remains one of the significant issues limiting its clinical application.
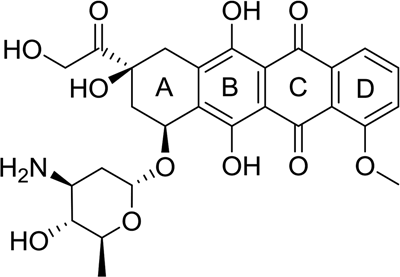
Figure 4 Molecular Structure of Doxorubicin
The mechanism of action of doxorubicin, like all anthracyclines, involves DNA intercalation. The planar aromatic system formed by rings B, C, and D can intercalate between the bases of the DNA double helix, stabilizing the complex through hydrogen bonds, hydrophobic interactions, and van der Waals forces, along with various functional groups and rings of adjacent DNA bases. Notably, the amino group in the sugar ring is believed to play a crucial role in the specific recognition of DNA molecules, which may cause doxorubicin to preferentially bind to guanine-rich regions of DNA. By forming tight DNA complexes, doxorubicin can inhibit the functions of DNA and RNA polymerases, leading to the cessation of DNA replication and RNA transcription, and blocking topoisomerases I and II.
The most prominent adverse effect associated with doxorubicin is cardiac toxicity. Studies have found that NADPH/CYP450 reductase reduction of the quinone C ring can generate reactive oxygen species (ROS), which further produce hydrogen peroxide (H2O2) through reactions catalyzed by superoxide dismutase. Ultimately, in the presence of ferrous ions, these can be converted into highly toxic hydroxyl radicals via the Fenton reaction. In the body, hydrogen peroxide can be converted to water and oxygen by catalase. However, this enzyme is lacking in cardiac tissue, making it particularly susceptible to damage from these free radical species. Furthermore, although doxorubicin cannot cross the blood-brain barrier, it can stimulate the production of tumor necrosis factor α (TNF-α), inducing inflammatory cytokines in microglia in the brain, which can then damage brain cells.
To mitigate the adverse effects of doxorubicin, researchers have encapsulated doxorubicin in liposomes. The resulting nanoparticle drug delivery system can extravasate in the loose and irregular blood vessels around the tumor tissue but cannot penetrate the myocardial area with tightly connected capillary junctions. This strategy, utilizing the enhanced permeability and retention effect, can effectively reduce the incidence of adverse reactions. However, one issue faced by liposome formulations is that once their surface is modified with opsonins (proteins that enhance phagocytosis), they are rapidly cleared from the blood by the reticuloendothelial system (RES). To counteract this phenomenon and extend their half-life, polyethylene glycol (PEG) can be used to coat liposomes, such as Doxil®. Myocet® and Nudoxa® utilize novel non-PEGylated liposomes to further improve the safety of the formulations.
To reduce doxorubicin toxicity, especially cardiac toxicity, it has also been introduced as a payload in ADCs. One of the most famous examples is the antibody-drug conjugate BR96-DOX, based on the chimeric antibody BR96. This antibody can recognize and bind to the Lewis-Y (Ley) epitope, which is highly expressed on the surface of most breast, colon, and lung cancer cells. Although this antigen is not exclusively present on tumor cells, BR96 has advantages over other antibodies targeting Lewis-Y, as it can be rapidly internalized and is more tumor-selective. This antibody conjugate is linked to doxorubicin via an acid-labile hydrazone bond, while the other end is conjugated to the BR96 antibody via an electrophilic maleimide group. After internalization of BR96–DOX and transfer to the acidic environment of the endosome, the hydrazone linker breaks, releasing the cytotoxic payload.
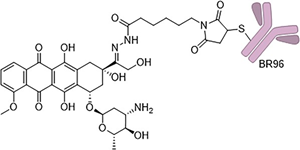
Figure 5 Molecular Structure of BR96–DOX, which conjugates doxorubicin to the Lewis Y-targeting antibody BR96 through an acid-labile hydrazone linker.
Although BR96-DOX exhibited significantly lower cytotoxicity in colon cancer cell lines (such as RCA and BN7005) compared to unconjugated doxorubicin, due to the presence of antigen-specific toxicity, it exhibited greater overall cytotoxicity than simply mixing the antibody and doxorubicin. In xenograft studies in athymic mice bearing human colon cancer (RCA), the cure rate was 89%, with a tumor regression rate of 12%, while unconjugated small molecule doxorubicin showed no significant effect at its maximum tolerated dose (MTD). Moreover, at this dose, the ADC did not cause any toxicity to the animals.
Another example of an ADC based on doxorubicin is Milatuzumab-doxorubicin (also known as IMMU-110 or hLL1-DOX), developed by Immunomedics, primarily targeting chronic lymphocytic leukemia (CLL) and non-Hodgkin lymphoma (NHL), which is currently in phase I/II trials (Figure 6). The target of this ADC, CD74, is a type II transmembrane glycoprotein associated with hematological malignancies but is also expressed in some solid tumors. Compared to antibodies against CD19 and CD22, anti-CD74 antibodies internalize much faster (approximately 100 times faster). It is estimated that CD74-expressing cells can internalize and degrade approximately 8 × 106 antibody molecules daily. Like previous ADCs, doxorubicin is also conjugated via an acid-labile hydrazone bond to 4-(maleimido methyl) cyclohexane-1-carboxylic acid hydrazone (MMCCH), with the other end of the linker conjugated to the antibody through the maleimide portion. In in vivo studies, the pharmacokinetic characteristics of IMMU-110 were found to be consistent with those of the naked antibody. The only difference was that the ADC had higher renal uptake; however, no acute renal toxicity or abnormal urinalysis results were observed in immunodeficient (SCID) mice and cynomolgus monkeys. In SCID mice, under a dose of 2.5 mg per mouse (equivalent to a dose of 3.6 mg/kg of doxorubicin), no toxicity was shown, whereas free doxorubicin had a maximum tolerated dose (MTD) of only 3 mg/kg in the same mouse model. The MTD for non-human primates (i.e., cynomolgus monkeys) was 30 mg/kg, with initial signs of bone marrow toxicity appearing with increased doses. However, even at doses as high as 90 mg/kg, no acute cardiac toxicity or toxicity to other major organs was observed. These preclinical safety and efficacy data suggest that IMMU-110 is a promising candidate for treating cancers with high CD74 expression (such as CLL, NHL, and multiple myeloma).
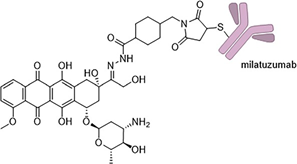
Figure 6 Structure of Milatuzumab-Doxorubicin IMMU-110, which is conjugated to the doxorubicin payload via an acid-labile hydrazone linker through the CD74-targeting antibody Milatuzumab.
Encouraging results have also been observed when doxorubicin is conjugated to trastuzumab. Trastuzumab targets the transmembrane protein human epidermal growth factor receptor 2 (HER2), which is overexpressed in certain types of breast cancer, and is internalized after antibody binding. The antibody-drug conjugate T-DOX developed by Zhang et al. (Figure 7) also consists of doxorubicin and MMCCH structure. In in vitro studies, this ADC retained selectivity for HER2-expressing cells while exhibiting lower cytotoxic effects on human cardiomyocytes compared to free doxorubicin. However, no results have yet indicated whether T-DOX has in vivo activity, and this ADC has not entered clinical trials.
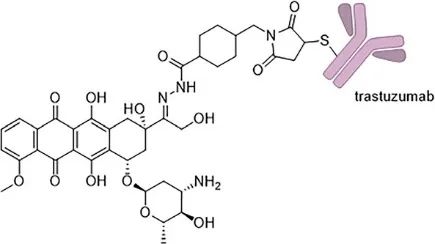
Figure 7 Structure of Trastuzumab-Doxorubicin Antibody-Drug Conjugate T-DOX, where doxorubicin is conjugated to the trastuzumab antibody via an acid-labile hydrazone bond.
Additionally, doxorubicin can also bind to epitope fragments of antibodies, increasing tumor permeability by reducing the molecular weight of the conjugate. Meanwhile, nanoparticles composed of polyamide-amine dendrimers (PAPAM) have been bifunctionalized to carry doxorubicin and antibodies into the tumor environment, utilizing the EPR effect to enhance tumor selectivity.
Methotrexate (MTX)
Since the mid-20th century, folate derivatives have been extensively studied and explored for clinical applications as effective antiproliferative agents. Through slight modifications of the folate structure, two antimetabolites were obtained: aminopterin (now commonly used as a rodenticide) and methotrexate (MTX) (Figure 8). Since 1953, MTX has been used to treat malignant tumors, including acute lymphoblastic leukemia, choriocarcinoma, non-Hodgkin lymphoma, and some solid tumors. Since 1958, it has also been used to treat psoriasis and was approved by the FDA for rheumatoid diseases in 1988. Its structure is derived from folate, with the hydrogen atom at position 4 of folate replaced by an amino group and the hydrogen atom at position 10 replaced by a methyl group. These modifications enhance MTX’s binding affinity to the active site of the DHFR enzyme by 104 times compared to the natural substrate.
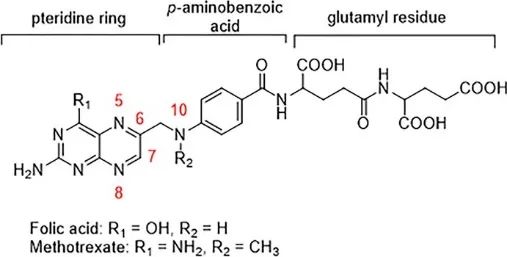
Figure 8 Structure of Folate and Methotrexate, showing their close structural similarity.
Methotrexate is a folate antagonist that can bind with high affinity to the DHFR site, which is the action site for the natural substrate 7,8-dihydrofolate (DHF). By strongly binding to DHFR, methotrexate inhibits the conversion of dihydrofolate to tetrahydrofolate, as tetrahydrofolate is an important cofactor in the conversion of 2-deoxyuridine monophosphate to thymidine monophosphate, leading to a decrease in thymidine levels and subsequently blocking DNA synthesis, resulting in cell death. Meanwhile, tetrahydrofolate is also crucial for the synthesis of inosine monophosphate, the conversion of serine to glycine, and the conversion of homocysteine to methionine. Therefore, methotrexate disrupts RNA and protein synthesis.
Like many other cytotoxic chemotherapeutic agents, methotrexate exerts toxic effects on tissues with high cell turnover rates. For instance, bone marrow, gastrointestinal mucosa, and hair follicles are particularly vulnerable. High-dose treatment regimens, such as when plasma concentrations exceed 10−4 M, result in a high incidence of bone marrow suppression (approximately 20% of patients), with 5% of patients experiencing severe bone marrow suppression. Furthermore, methotrexate can cause mucositis, hepatotoxicity, acute renal failure, hair loss, rash, and other adverse reactions. To avoid these adverse reactions, monitoring plasma concentrations of methotrexate during administration is necessary, and when plasma concentrations exceed 10−6 M and persist for 24 hours, leucovorin should be administered. Leucovorin can act as a cofactor in reactions requiring tetrahydrofolate without going through the DHFR pathway.
In recent years, various prodrugs and antibody-drug conjugates based on MTX have been developed (Figure 9). In early work, the combination of MTX with immunoglobulin G (IgG) antibodies improved drug specificity but reduced drug efficacy and the ability of the antibody to bind to the target. Scientists subsequently developed improved methotrexate conjugates that exhibited greater potency than standalone methotrexate while maintaining almost no loss in antibody binding ability. Additionally, there have been reports on using peptides, enzymes, and other carrier systems (such as serum albumin) to conjugate MTX.
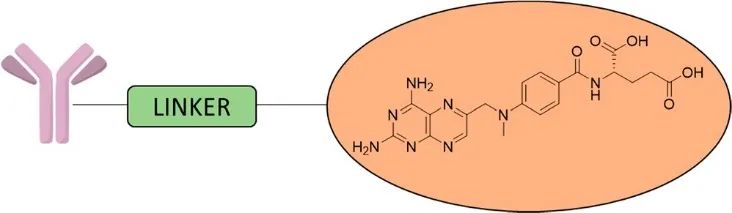
Figure 9 Schematic Diagram of MTX-ADC Structure
Other Traditional Cytotoxic Drugs
To date, many ADCs based on cytotoxic drugs have been developed, but many have not been evaluated clinically. For example, platinum drugs (such as cisplatin, Figure 10a) are widely used to treat malignant tumors but can lead to severe side effects. To overcome this issue, scientists have produced an ADC composed of Herceptin and platinum-based drugs, where the protein is linked to the small molecule drug through a dipeptide linker that can be cleaved by tissue protease B. From the IC50 data of three breast cancer cell lines (SKBR-3, MDA-MB-231, and MCF-7), the Herceptin–Pt(II) conjugate exhibited specific cytotoxicity against HER2-positive cancer cell lines after co-culturing with the cells for 72 hours (i.e., IC50=19.7µM in SKBR3, but negligible activity in the HER2-negative cell line MCF-7). Platinum compounds have also been utilized in antibody-nanoparticle conjugates. For instance, the Ahn research group utilized nanoparticle carriers to deliver platinum-based drugs to tumor cells. These conjugates demonstrated significant cell binding and rapid internalization, exhibiting strong in vitro cytotoxicity (after 48 hours of culture). Additionally, in vivo experiments indicated that the growth of pancreatic tumor xenografts was inhibited for over 40 days, outperforming small molecule drugs at equivalent concentrations.
Paclitaxel
Paclitaxel (Figure 10b) can also serve as an effective payload in ADCs. As a mitotic inhibitor, paclitaxel can prevent excessive division of tumor cells. The Guillemard research group first reported the synthesis of paclitaxel-antibody conjugates and subsequently conducted a study with long-circulating liposomes conjugated to fully human anti-vascular endothelial growth factor (VEGF) monoclonal antibodies. The PEGylated liposomes loaded with paclitaxel were conjugated to the antibodies through a maleimide (Mal)-PEG2000-DSPE linker, generating stable VEGF mAb liposomes. An antitumor activity evaluation model was established in BALB/c nude mice bearing SGC-7901 xenografts. The results indicated that mice treated with monoclonal antibody-liposome paclitaxel exhibited superior antitumor effects compared to small molecule paclitaxel and liposome controls after five intravenous injections.
Another study utilized nanoparticles to investigate the role of paclitaxel in ADC conjugates, where an anti-hypoxia-inducible factor (HIF)-1a antibody was conjugated to single-molecule polymeric micelles filled with paclitaxel. After 48 hours of incubation, the efficacy of this conjugate was evaluated in gastric cancer MGC-803 cells and HDF fibroblasts. The results showed that this nanoparticle drug selectively killed MGC-803 cancer cells.
Paclitaxel has also been conjugated with a peptide that recognizes a specific receptor, which increased cytotoxicity against H1299 human non-small cell lung cancer cells compared to using paclitaxel alone. The same research group also developed a paclitaxel-based ADC using the anti-epidermal growth factor receptor (EGFR) monoclonal antibody cetuximab. In vitro data from A431, UM-SCC-1, and UM-SCC-6 cells showed that the cytotoxicity of the paclitaxel-based ADC was increased compared to free drug, intact antibody, or physical mixtures of both. However, the cytotoxicity data was not supported by in vivo results, where no significant differences were observed between the ADC and free paclitaxel in animal models. The reason for this result may relate to the dosing issues of the ADC or the premature release of the ADC payload.
Dacarbazine
Modified surface nanoparticles loaded with dacarbazine (DTIC) have also been conjugated to antibodies, showing promising results (Figure 10c). Dacarbazine induces apoptosis by methylating DNA bases and is commonly used to treat melanoma, and it can also be used in combination with other drugs for treating Hodgkin’s disease, Kaposi’s sarcoma, neuroblastoma, and various other cancers. In these studies, dacarbazine was loaded into polylactic acid (PLA) nanoparticles and then covalently conjugated to the anti-tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-receptor 2 (DR5) antibody. The conjugate exhibited better efficacy compared to equal amounts of dacarbazine or dacarbazine nanoparticles used alone. Simultaneously, no cytotoxicity was observed in NIH mouse fibroblast cell lines with either DTIC nanoparticles or DTIC-DR5 mAb nanoparticles.
Vincristine
Vincristine can also be applied in ADCs. The Yurkovetskiy research group investigated polymer-based antibody-vincristine conjugates. This study utilized a polymer ADC platform made from poly(1-hydroxymethyl vinyl-1-hydroxymethyl formaldehyde) (PHF, also known as “Fleximer”), which significantly improves the hydrophilicity, allowing for a higher number of effective payloads in the ADC compared to other conventional ADCs, producing conjugates with a drug-to-antibody ratio (DAR) of up to 20. Scientists conducted in vitro evaluations of the unconjugated PHF-vincristine and the small molecule vincristine compound N-(3-hydroxypropyl) vincristine in HER2-positive and negative cell lines. The results showed that the ADC (DAR=20) exhibited potency in HER2-negative cell lines (MCF7) that was 10 to 15 times lower than in positive cell lines.
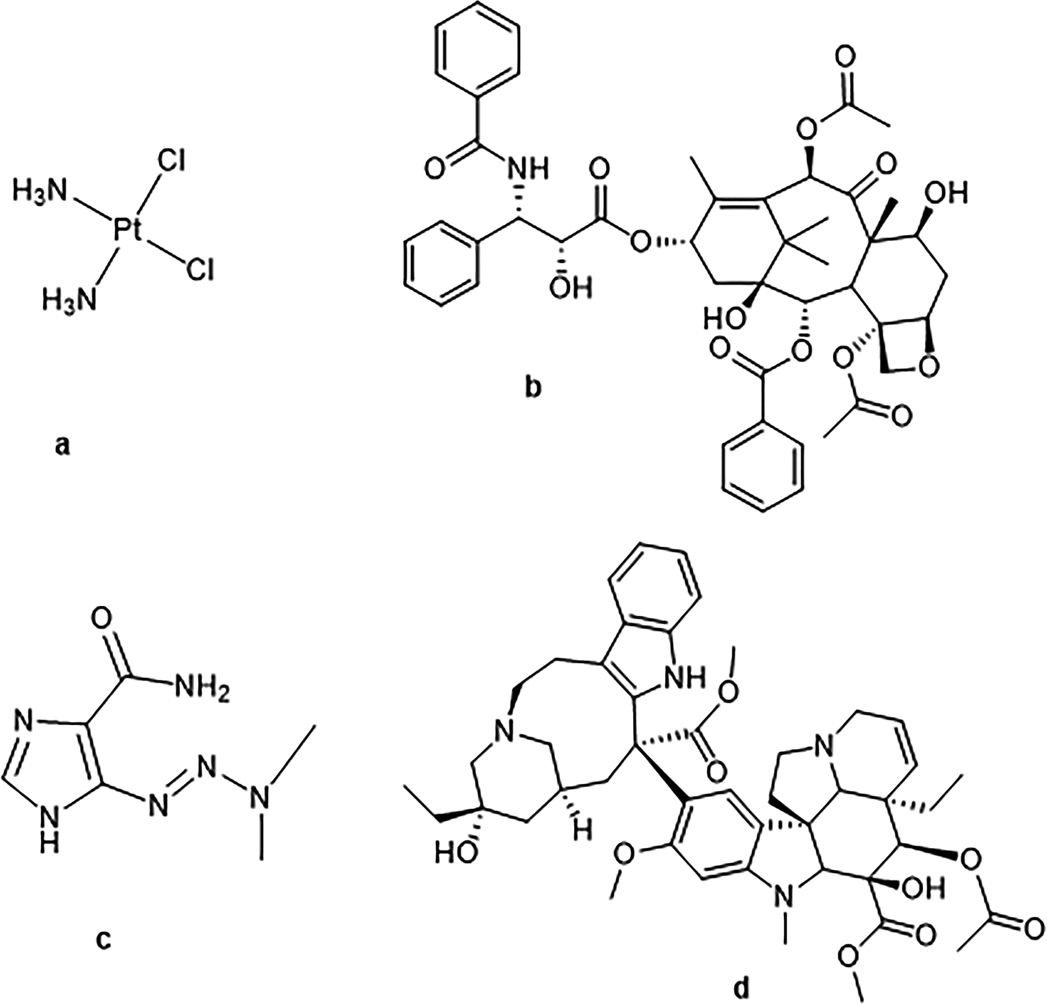
Figure 10 Structures of Various Cytotoxic Drugs: (a) Cisplatin (b) Paclitaxel (c) Dacarbazine and (d) Vincristine
-
Conclusion
Doxorubicin, mitomycin C, and methotrexate have milestone significance as independent anticancer drugs in the field of cancer treatment. Therefore, it is understandable that they are preferred as ADC payloads. However, as of now, no ADC using these payloads has been approved. The most recent example is BR96-DOX based on doxorubicin, which was tested in phase II clinical trials for treating breast cancer but was terminated due to failure to demonstrate effective therapeutic activity.
Although they did not ultimately reach the market, these pioneering attempts have furthered our understanding of the mechanisms of action of ADCs and their safe and effective pharmacological characteristics.
First, due to various issues arising from murine antibodies, there is a recognition of the importance of using humanized or chimeric antibodies to avoid immune reactions.
Second, it has been found that maintaining the stability of linkers during circulation is crucial when using cleavable linkers, as this can prevent premature release of the payload, leading to unnecessary systemic toxicity and reducing the therapeutic index.
Finally, a key issue with first-generation ADCs is the insufficient cellular uptake of the payload. In some cases, the efficacy of the conjugate is significantly lower than that of the parent free drug, which can be attributed to the different cellular uptake patterns of the conjugate compared to free small molecules. Typically, there are about 10^5 antigen molecules per cell surface, while for moderately potent cytotoxic drugs like doxorubicin and mitomycin C, the number of molecules required to produce an effect is estimated to be 10^6 or more per cell.
In summary, these observations and conclusions are crucial for the later development of new and more effective payloads, improved linkers, and the selection of appropriate antibodies. These studies have also contributed to the approval of ADCs such as Kadcyla®, Mylotarg®, Adcetris®, Besponsa®, and immunotoxins like Lumoxiti® and Elzonris®.