CK Note: The diagnosis and treatment of primary aldosteronism have formed a system, but the relatively complex processes and controversial results have left many endocrinologists confused. Primary aldosteronism also faces many challenges, mainly due to the high prevalence that exceeds previous understandings and the simplification of processes and methods; all of these require evidence. Current research in this field should follow the trend. This series focuses on the current situation and challenges.This series is based on a literature review of primary aldosteronism conducted in early 2021, with expansions…
The first four parts of this series can be viewed:
-
Clinical Practice | Diagnosis and Treatment of Primary Aldosteronism: Reality and Challenges Part One – Public Health Issues?
-
Clinical Practice | Diagnosis and Treatment of Primary Aldosteronism: Reality and Challenges Part Two – Unification of Genetics and Pathophysiology
Clinical Practice | Diagnosis and Treatment of Primary Aldosteronism: Reality and Challenges Part Three – The Organogenesis and Biomarkers (APCC and APA) of Aldosteronism
-
Clinical Practice | Diagnosis and Treatment of Primary Aldosteronism: Reality and Challenges Part Four – Screening and Diagnosis?
Clinical Practice | 2021 | Realities and Challenges
Diagnosis and Treatment of Primary Aldosteronism
Subtyping of Primary Aldosteronism
Compiled by Chen Kang
The purpose of subtype classification is to identify suitable candidates for unilateral adrenalectomy, i.e., patients who can achieve biochemical remission postoperatively with a low risk of recurrence. The two subtypes of primary aldosteronism—unilateral and bilateral—represent extremes of morphological and biochemical phenotype spectra, rather than being completely distinct. In other words, asymmetrical bilateral aldosteronism with partial lateralized secretion is common, while true unilateral aldosteronism without contralateral aldosterone secretion is rare. Some demographic, clinical, biochemical, and imaging characteristics are associated with subtypes of primary aldosteronism.CT and MRI Imaging of the AdrenalsAdrenal CT is the first step in assessing the subtypes of primary aldosteronism and should be performed in every patient if possible[J Clin Endocrinol Metab 2016; 101: 1889–916; J Hypertens 2020; 38: 1919–28]. The imaging features of the adrenals in patients with primary aldosteronism may include[J Clin Endocrinol Metab 2017; 102: 1182–92]:
- Adrenals with no structural change,
- Unilateral adenoma,
- Bilateral adenomas,
- Small nodular hyperplasia,
- Bilateral large nodular hyperplasia
- (rare) Unilateral adrenal cortical carcinoma.
The CT imaging phenotype may also provide indications for further treatment, such as the following situations:
- Adrenal cortical carcinoma (rare);
- Some younger patients with unilateral adenoma may bypass AVS, with the next step being unilateral adrenalectomy (Figure 14).
- Some patients with bilateral large nodular hyperplasia may bypass AVS, as the likelihood of bilateral aldosteronism is higher.
- For patients with concomitant adrenal (clinical) Cushing’s syndrome and unilateral masses, AVS may be bypassed, and initial treatment should aim at resecting the adenoma producing cortisol.
Figure 14 Simplified Pathway for Further Typing Treatment of Diagnosed Aldosteronism(Source: 14th Edition of Williams Endocrinology, Mayo)
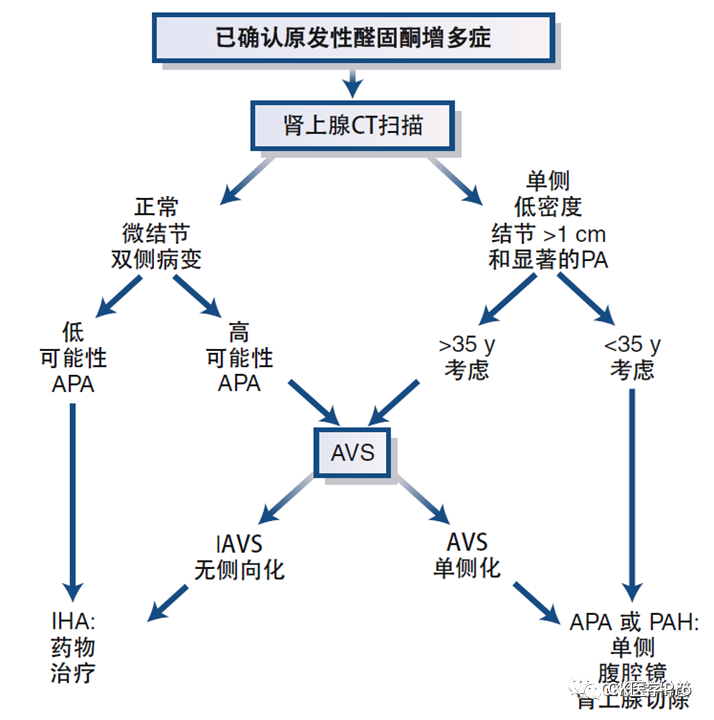
Interventional radiologists should carefully examine imaging to better plan AVS. CT usually does not provide information on the secretory activity of detected nodules[AJR Am J Roentgenol 2010; 194: 1450–60]. Similarly, density measurements (CT values/Hounsfield units) and parameters of contrast agent clearance cannot differentiate between secretory and non-secretory nodules[J Clin Endocrinol Metab 2014; 99: E1035–39; Lancet Diabetes Endocrinol 2015; 3: 296–303; J Clin Endocrinol Metab 2016; 101: 1889–916.].As adrenal adenomas become increasingly common with age[Lancet Diabetes Endocrinol 2020; 8: 894–902], the accuracy of CT imaging for subtype classification of primary aldosteronism is lower in older patients[J Clin Endocrinol Metab 2014; 99: 2712–19; Ann Intern Med 2009; 151: 329–37]. Overall, in approximately 40% of patients with primary aldosteronism, adrenal CT imaging is inconsistent with AVS results[J Clin Endocrinol Metab 2014; 99: 2712–19; J Hypertens 2021; 39: 310–17].Conversely, as adrenal adenomas are not common in younger individuals, such patients with primary aldosteronism and unilateral small adenomas (typically <2 cm) usually indicate unilateral disease[Lancet Diabetes Endocrinol 2020; 8: 894–902]. The accuracy of CT imaging in determining the subtype of primary aldosteronism is highest in patients under 35, but it is not perfect[J Clin Endocrinol Metab 2014; 99: 2712–19; Clin Endocrinol (Oxf) 2018; 88: 645–51].Preoperative CT scans for AVS also help locate the adrenal veins. MRI is not as good as CT due to its lower spatial resolution and is therefore the second choice for imaging[J Clin Endocrinol Metab 2016; 101: 1889–916; J Hypertens 2020; 38: 1929–36].Adrenal Venous SamplingAVS is a technically challenging, expensive, and poorly standardized procedure, with significant differences in success rates between centers, depending on the expertise of the interventional radiologist. In experienced centers, the technical success rate of AVS can reach 90% or higher[Clin Endocrinol (Oxf) 2009; 70: 14–17; Hypertension 2014; 63: 151–60].Many studies supporting the use of AVS are based on low-quality evidence, including retrospective designs, no controls, and no clinical endpoints (such as biochemical remission). Despite these limitations, AVS is consistently used worldwide[J Clin Endocrinol Metab 2012; 97: 1606–14]. Only one prospective randomized trial, SPARTACUS, compared treatment decisions based on AVS and CT in 184 patients, finding no difference in postoperative blood pressure outcomes[Lancet Diabetes Endocrinol 2016; 4: 739–46]. However, this study has been widely discussed and criticized due to its overall study design and potential lack of power[Hypertension 2017; 69: 396–97]. A multicenter international study using a non-randomized retrospective approach supported the biochemical remission rates of the strategies in the SPARTACUS trial: 188 of 235 patients (80%) achieved biochemical remission after CT-based treatment decisions, while 491 of 526 patients (93%) achieved biochemical remission after AVS-based treatment decisions (p < 0.001)[Hypertension 2018; 72: 641–49]. AVS is currently still recommended as the gold standard for subtype classification; however, there are many controversies (Table 10), one major controversy is how to determine the applicable population for AVS, considering its complexity and cost-effectiveness, especially as primary aldosteronism is increasingly recognized as common (the current consensus on the range of use is seen in Table 11).Table 10 Controversies or Areas Needing Further Research Regarding AVS
-
Applicable population for AVS
- Whether AVS can be replaced
-
Preoperative preparation for AVS
-
AVS procedural pathways
-
Whether AVS uses ACTH
-
Interpretation of AVS data
Table 11 Current Consensus Recommended Scope of Use for AVS

According to an international study, 46% of major centers use adrenocorticotropic hormone (1-24) infusion during AVS[J Clin Endocrinol Metab 2012; 97: 1606–14]. Arguments supporting the use of adrenocorticotropic hormone (1-24) include reducing stress-induced changes in aldosterone secretion, improving the technical success rate of AVS, and maximizing aldosterone secretion from the adenoma producing aldosterone. Arguments against this method include reducing the proportion of lateralized AVS results, thus reducing the proportion of patients who are operable[J Clin Endocrinol Metab 2020; 105: dgz017.]. Some studies compared AVS results before and after adrenocorticotropic hormone (1-24) stimulation, reporting inconsistent results in up to 25% of patients. During AVS, particularly when not using adrenocorticotropic hormone (1-24), detailed steroid analysis beyond cortisol and aldosterone is considered to have potential value[Clin Chem 2016; 62: 514–24; Hypertension 2020; 75: 183–92].AVS is performed via a percutaneous femoral vein approach. The adrenal veins can be cannulated sequentially bilaterally or simultaneously bilaterally[AJR Am J Roentgenol 2010; 194: 1450–60]. Blood is slowly aspirated from the left adrenal vein (which drains directly into the left renal vein) and the right adrenal vein (which typically drains directly into the inferior vena cava and is difficult to locate)[Lancet Diabetes Endocrinol 2015; 3: 296–303]. The ratio of cortisol in each adrenal vein to cortisol in peripheral veins needs to be sufficiently high to verify whether AVS is technically successful (Table 12 and Table 13). Rapid cortisol testing can be used to improve technical success rates because if the results do not support successful cannulation, it allows for resampling of the adrenal vein[Ann Surg 2009; 249: 318–21; Eur J Endocrinol 2011; 165: 301–06]. The AVS index is calculated based on the detection values of cortisol and aldosterone in the adrenal and inferior vena cava veins (Table 12 and Table 13). Complications of AVS include the most common inguinal hematoma, and rare adrenal hemorrhage or adrenal vein dissection, with an overall complication rate of about 2% in experienced AVS centers[Clin Endocrinol (Oxf) 2009; 70: 14–17; Hypertension 2016; 67: 146–52; Radiographics 2005; 25 (suppl 1): S143–58].Table 12: Comparison of AVS Guidelines for Patients with Aldosteronism
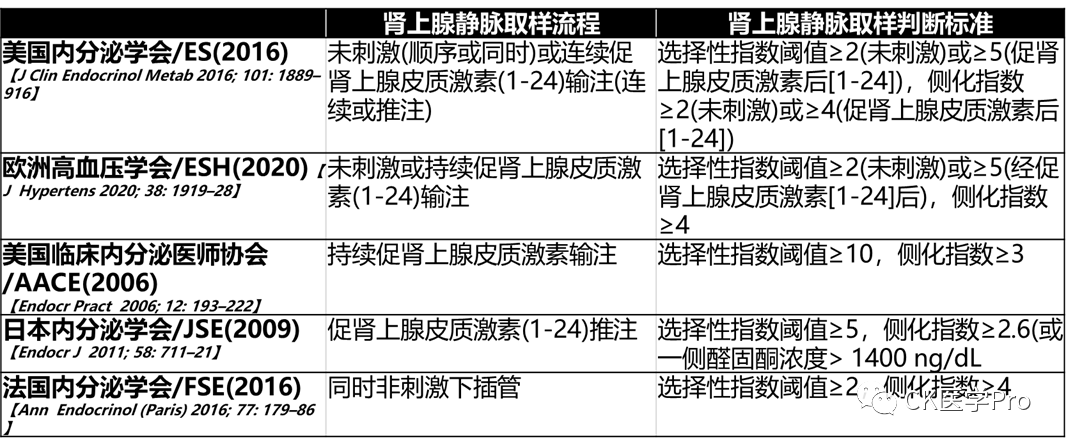
Table 13 Correct Interpretation Steps for Adrenal Vein Sampling in Patients with Aldosteronism with and without Adrenocorticotropic Hormone (1-24) StimulationAdrenal Venous Sampling

Correct interpretation of adrenal venous sampling results requires several steps. If the diagnosis is correct and the case screening is positive, AVS can be performed regardless of what medications are used. For example, in patients treated with low doses of mineralocorticoid receptor antagonists (such as spironolactone or eplerenone), if plasma renin activity remains suppressed, AVS can be performed[J Clin Endocrinol Metab 2019; 104: 487–92]. Co-secretion of cortisol is common in patients with primary aldosteronism and is mild in most cases[JCI Insight 2017; 2: 93136]. When patients with co-secretion of primary aldosteronism are diagnosed with (obvious) Cushing’s syndrome (non-ACTH dependent), generally, the surgical target is the excess cortisol. In these cases, AVS may not be necessary, either because the treatment target is to remove the adrenal gland to treat cortisol excess (i.e., when there is a unilateral adrenal mass) or because there may be a bilateral process (i.e., when imaging shows large nodular hyperplasia). In mild autonomous cortisol secretion, if performed during adrenocorticotropic hormone (1-24) stimulation, the interpretation of AVS seems to have little impact[J Clin Endocrinol Metab 2020; 105: dgaa519].Figure 15 and Table 13 present the diagnostic process for subtyping using AVS and the standards for interpreting AVS results.Figure 15 Flowchart for Subtyping Primary Aldosteronism
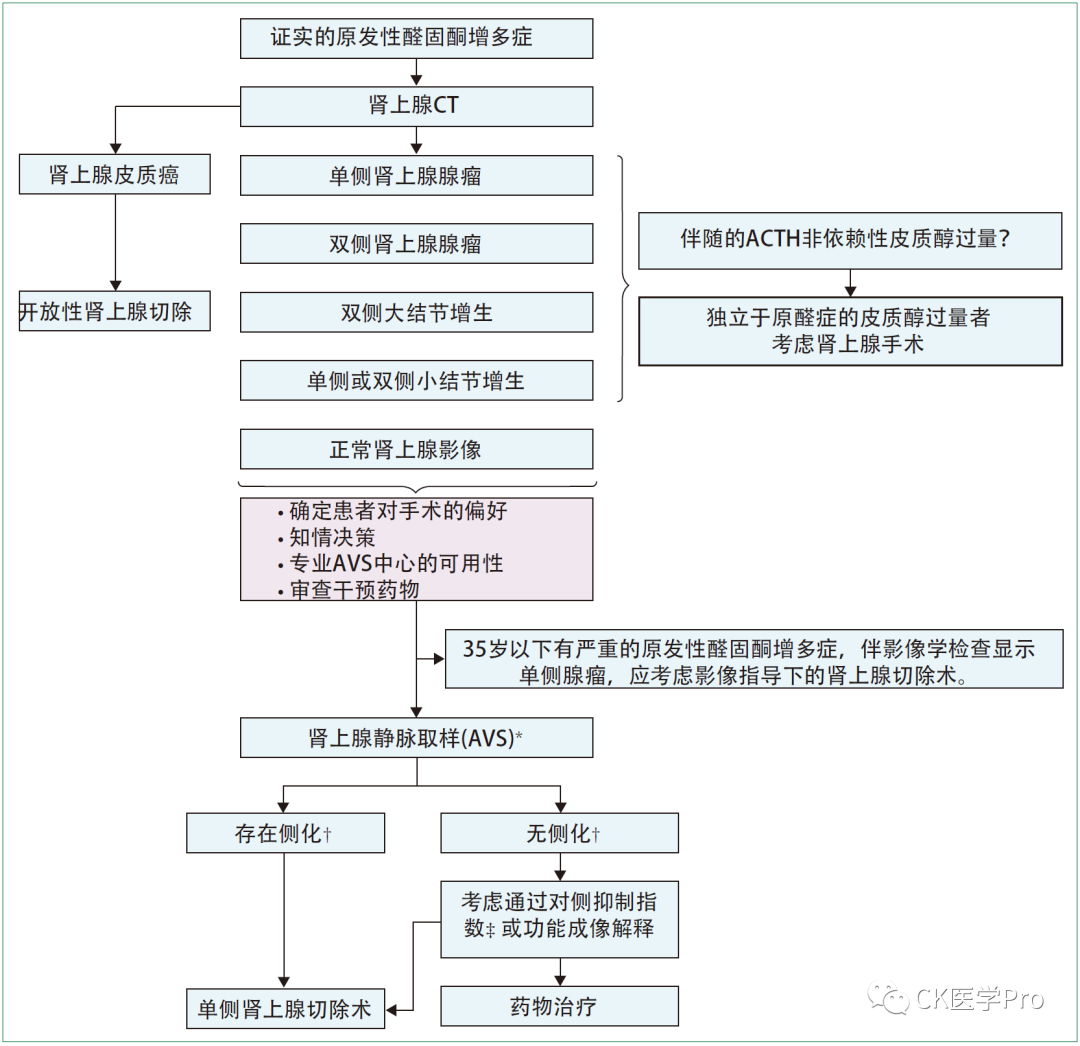
* Based on the selective index in Table 13.
† Based on the lateralization index in Table 13.
‡ Based on the contralateral suppression index in Table 13.
Lancet DE 2021
Reports have used predicted scores combining clinical, biochemical, and adrenal imaging parameters for the prediction of subtypes of primary aldosteronism (Table 14, recently reported scoring models). A simple application of scores in a large cohort of 1936 patients with primary aldosteronism (randomly assigned to development and validation datasets) performed well in predicting bilateral primary aldosteronism (positive predictive value of 93±5% for scores ≥8)[Clin Endocrinol (Oxf) 2018; 88: 645–51]. An additional predictive model developed in a smaller cohort of 150 patients is accessible through an online tool (sensitivity 91±7%; specificity 79±3%), and was validated in an independent external cohort[J Clin Endocrinol Metab 2020; 105: dgaa379]. Although predictive scores may have potential significance in bypassing AVS in patients classified as bilateral disease and guiding surgical decisions, some limitations must be considered, such as inter-center variability in aldosterone and renin assays, the heterogeneity of imaging techniques and their interpretation and reporting, and the scores not being derived from large multicenter cohorts, etc., which may limit their generalizability to other populations.Table 14 Scoring Models for Predicting Aldosteronism Subtypes

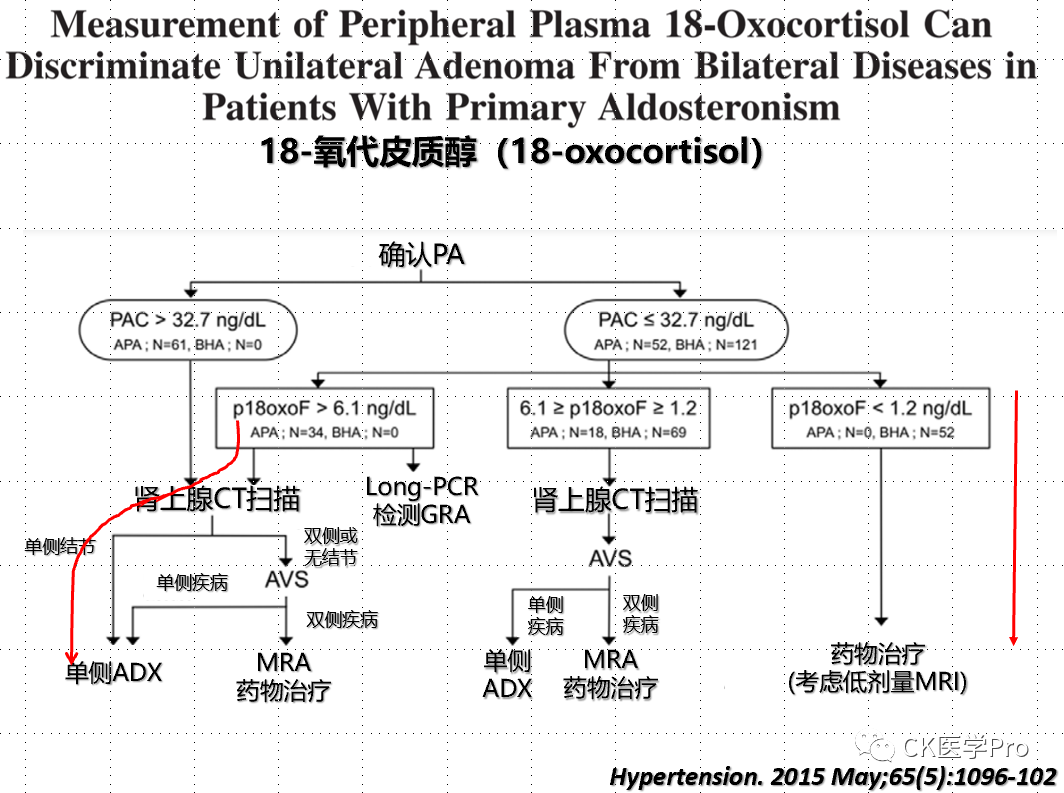
Steroid ProfilesSeveral studies have shown varying diagnostic accuracies when analyzing 18-hydroxycorticosterone and 18-hydroxycortisol in AVS samples to differentiate between unilateral and bilateral primary aldosteronism (Figure 16a,b,c)[Hypertension 2015; 65: 1096–102; Clin Chem 2016; 62: 514–24].Figure 16a,b,c The Value of 18-Hydroxycorticosterone, 18-Hydroxycortisol, and 18-Oxocorticosterone in Aldosteronism Subtyping(a)
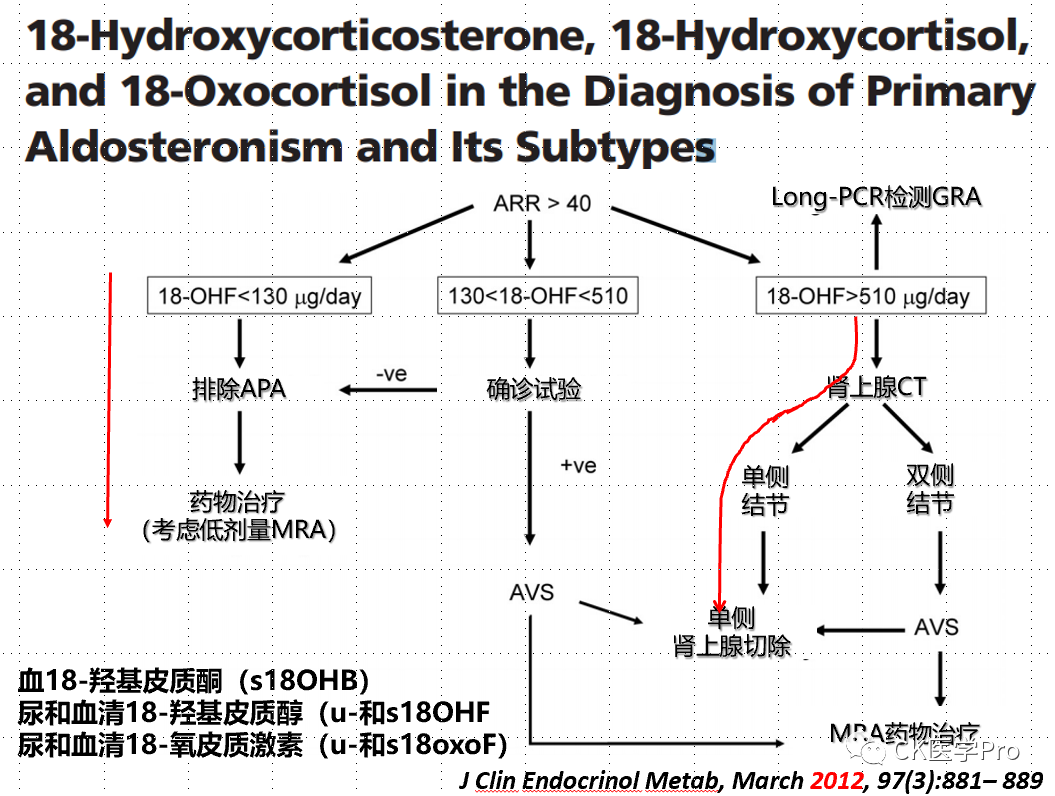
(b)
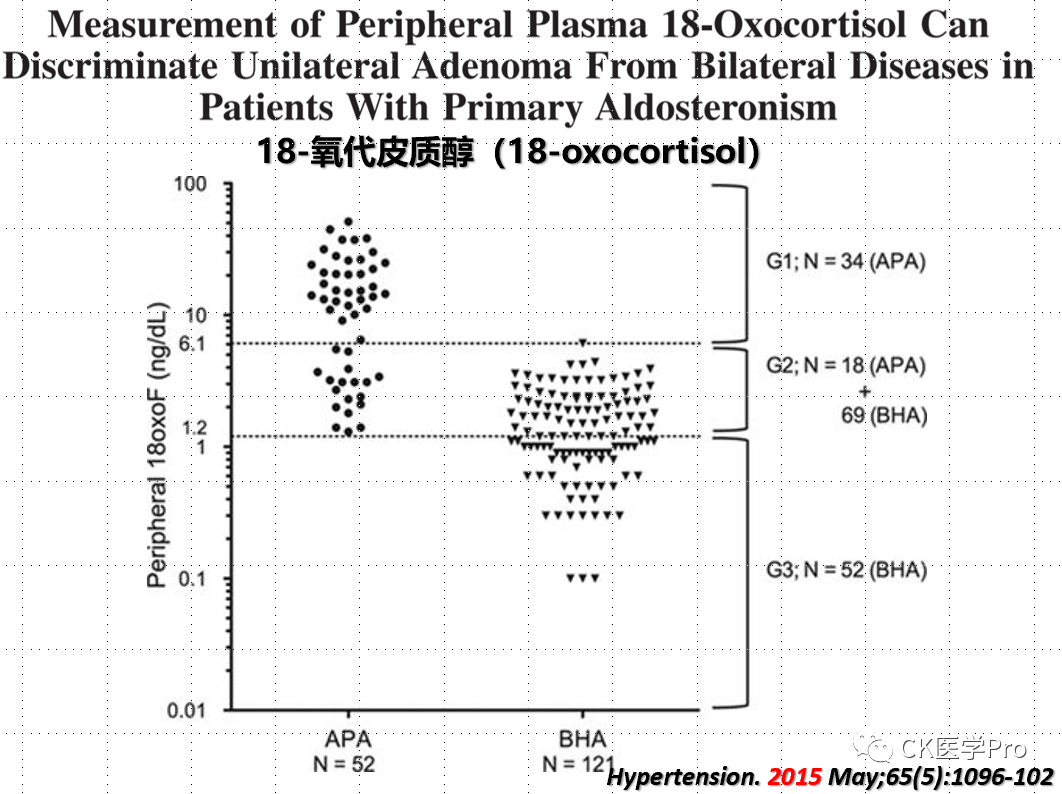
(c)
Additionally, serum and urine steroid metabolomics provide deeper elucidation of the biosynthesis, metabolism, and excretion of steroid hormones, and facilitate the revelation of potential enzymatic deficiencies related to steroidogenesis. Although changes in steroid metabolomics have been used to diagnose congenital disorders of steroidogenesis for decades, recent advancements have refocused attention on the analytical capabilities of steroid metabolomics. The combination of mass spectrometry-based steroid profiling with machine learning data analysis has created a powerful steroid metabolomics discovery tool, making it very suitable as a diagnostic biomarker (omics) method. Moreover, the development of new technologies has facilitated the advancement of high-throughput multi-steroid profiling methods, which will soon be applicable in clinical practice.
The adrenal cortex, gonads, and placenta are the primary sites of cholesterol metabolic synthesis (Figure 17). Some of the steroids produced can directly bind and activate steroid receptors in target cells, while others require downstream activation but can also be inactivated or redirected to other steroid metabolic pathways. This intracellular steroid receptor pre- and post-metabolism is also referred to as “intracrine (secretion within cells),” which explains why circulating steroid concentrations often do not represent the observed biological hormonal activity (for instance, the high concentrations of testosterone in the testes exceeding serum levels enable spermatogenesis). Furthermore, adrenal steroidogenesis exhibits circadian rhythms, thus single-time-point serum steroid measurements provide only immediate information. Quantitative analysis of total steroid net output in 24-hour urine can avoid this issue.
Figure 17 Diagram of Steroid Generation and Corresponding Urinary Steroid Metabolites
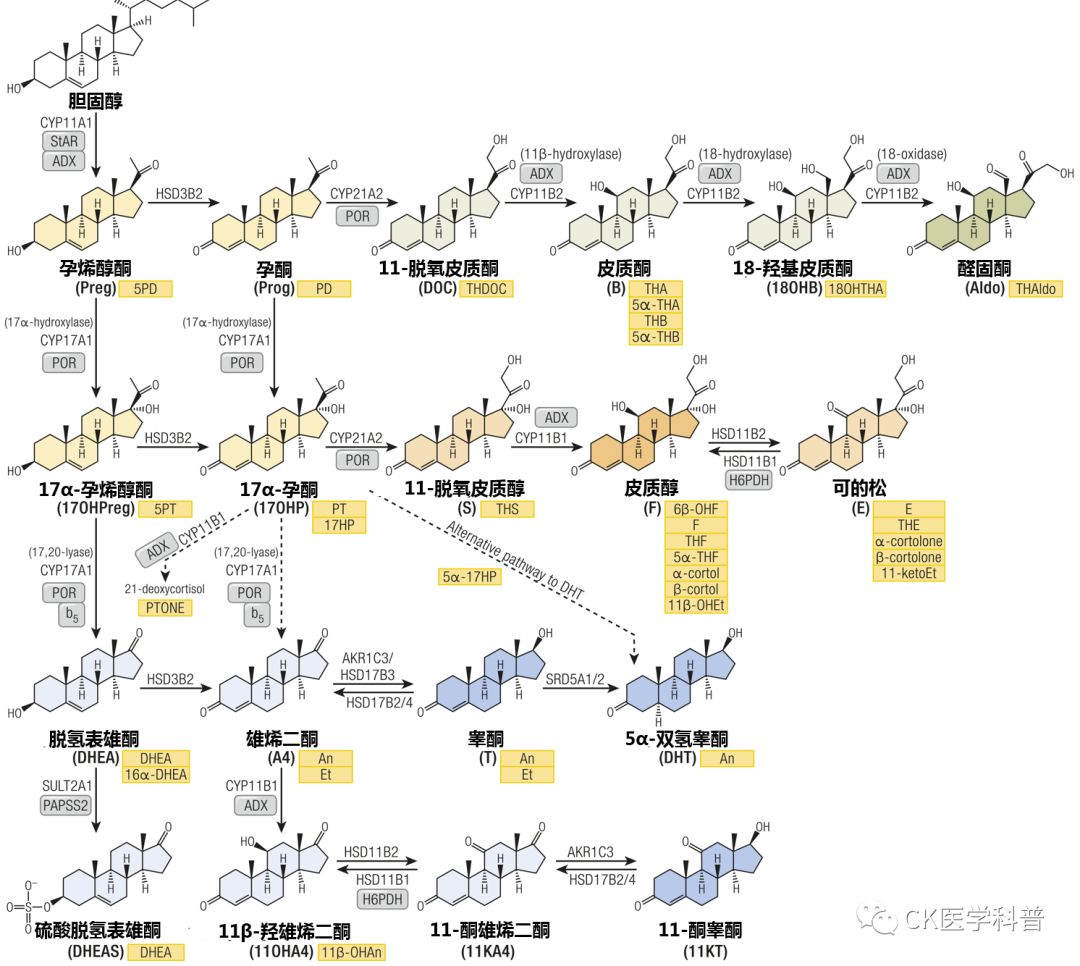
Steroids are color-coded based on their biological activity or contribution to specific pathways: general precursors (yellow), mineralocorticoid precursors (light green), active mineralocorticoid precursors (dark green), glucocorticoid precursors and inactive metabolites (light orange), active glucocorticoid precursors (dark orange), androgen precursors (light blue), and active androgens (dark blue). Corresponding urinary metabolites are shown in yellow boxes. Arrows indicate catalytic enzymes and isoforms. The diagram highlights essential cofactor proteins: ADX: adrenodoxin, adrenal cortical ferredoxin; b5: cytochrome b5; PAPSS2: PAPS synthase 2; PRO: cytochrome P450 oxidoreductase; StAR, steroidogenic acute regulatory protein.
Autonomous adrenal steroid excess steroid metabolomics currently have diagnostic value in ACTH-independent Cushing’s syndrome and ACTH-dependent Cushing’s syndrome, MACE (mild autonomous cortisol secretion), primary aldosteronism (Figure 18), adrenal cortical carcinoma (ACC), etc.Figure 18 The Value of Steroid Metabolomics in Aldosteronism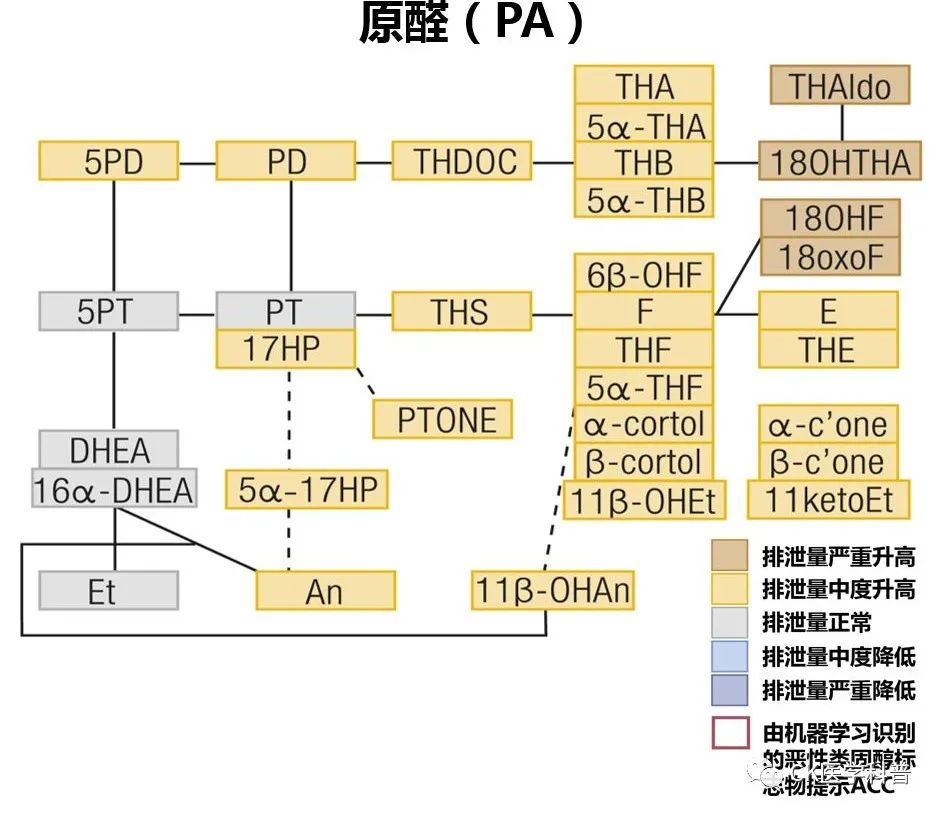 Using liquid chromatography-mass spectrometry-based methods to analyze 12 steroids in peripheral blood samples accurately identified 172 of 216 patients with primary aldosteronism (80%)[Clin Chem 2016; 62: 514–24.]. In another study, seven steroid fingerprint profiles correctly classified 73 of 79 patients with unilateral primary aldosteronism (92%) based on genotype[Hypertension 2016; 67: 139–45]; however, when tested in a larger patient cohort, steroid profiling was less accurate. In a large multicenter study involving patients with primary aldosteronism and hypertension, steroid analysis combined with machine learning algorithms diagnosed primary aldosteronism with a sensitivity of 69% and specificity of 94%, and identified primary aldosteronism associated with KCNJ5 gene variants, with a sensitivity of 85% and specificity of 97%[JAMA Netw Open 2020; 3: e2016209]. Steroid profiling is currently available only in a few centers and requires validation before being used in routine clinical management.Functional ImagingSeveral functional imaging techniques for subtyping primary aldosteronism have been explored (Figure 19). In some small studies, 11C-metomidate PET has been used for the diagnosis of primary aldosteronism[J Clin Endocrinol Metab 2012; 97: 100–09; Clin Endocrinol (Oxf) 2019; 90: 670–79]. However, this method has some challenges, such as the lower selectivity of 11C-metomidate for CYP11B2 (aldosterone synthase) compared to CYP11B1 (11β-hydroxylase), requiring dexamethasone pre-treatment, and the limited number of centers capable of detection due to the short half-life of C11. One study showed that the consistency of C-metomidate PET imaging with AVS results was only 51%, but not all patients underwent dexamethasone pre-treatment[Eur J Endocrinol 2020; 183: 539–50,Figure 19].Figure 19 Nuclear Medicine Imaging for Subtyping Primary Aldosteronism
Using liquid chromatography-mass spectrometry-based methods to analyze 12 steroids in peripheral blood samples accurately identified 172 of 216 patients with primary aldosteronism (80%)[Clin Chem 2016; 62: 514–24.]. In another study, seven steroid fingerprint profiles correctly classified 73 of 79 patients with unilateral primary aldosteronism (92%) based on genotype[Hypertension 2016; 67: 139–45]; however, when tested in a larger patient cohort, steroid profiling was less accurate. In a large multicenter study involving patients with primary aldosteronism and hypertension, steroid analysis combined with machine learning algorithms diagnosed primary aldosteronism with a sensitivity of 69% and specificity of 94%, and identified primary aldosteronism associated with KCNJ5 gene variants, with a sensitivity of 85% and specificity of 97%[JAMA Netw Open 2020; 3: e2016209]. Steroid profiling is currently available only in a few centers and requires validation before being used in routine clinical management.Functional ImagingSeveral functional imaging techniques for subtyping primary aldosteronism have been explored (Figure 19). In some small studies, 11C-metomidate PET has been used for the diagnosis of primary aldosteronism[J Clin Endocrinol Metab 2012; 97: 100–09; Clin Endocrinol (Oxf) 2019; 90: 670–79]. However, this method has some challenges, such as the lower selectivity of 11C-metomidate for CYP11B2 (aldosterone synthase) compared to CYP11B1 (11β-hydroxylase), requiring dexamethasone pre-treatment, and the limited number of centers capable of detection due to the short half-life of C11. One study showed that the consistency of C-metomidate PET imaging with AVS results was only 51%, but not all patients underwent dexamethasone pre-treatment[Eur J Endocrinol 2020; 183: 539–50,Figure 19].Figure 19 Nuclear Medicine Imaging for Subtyping Primary Aldosteronism

Additionally, high expression of CXC chemokine receptor type 4 (CXCR4) has been reported in aldosterone-producing adenomas and is associated with CYP11B2. The CXCR4 ligand 68Ga-pentixafor PET successfully detected primary aldosteronism in a preliminary small study involving 9 patients[Hypertension 2018; 71: 317–25]. In a reference standard study based on clinical follow-up and immunohistochemistry involving 36 patients, 68Ga-pentixafor PET achieved an efficiency of 88% sensitivity and 100% specificity for diagnosing aldosteronism[Eur J Nucl Med Mol Imaging 2020; 47: 2656–65]. Larger studies are needed to determine the optimal reference standard.
Familial Hyperaldosteronism (FH)
Information regarding familial hyperaldosteronism has been described in the aforementioned section on the mechanisms of primary aldosteronism.
Clinically, such patients are relatively rare; however, FH should be considered when PA is diagnosed before the age of 20 or when PA is diagnosed in more than one family member.
So far, four forms of FH have been described[Eur J Endocrinol 2018; 178: R101–11]. However, it is likely that more pathogenic germline mutations will be described in the coming years. A more practical naming method is to use the specific gene mutations that are pathogenic (see Table 15).
Table 15 Forms of Familial Hyperaldosteronism
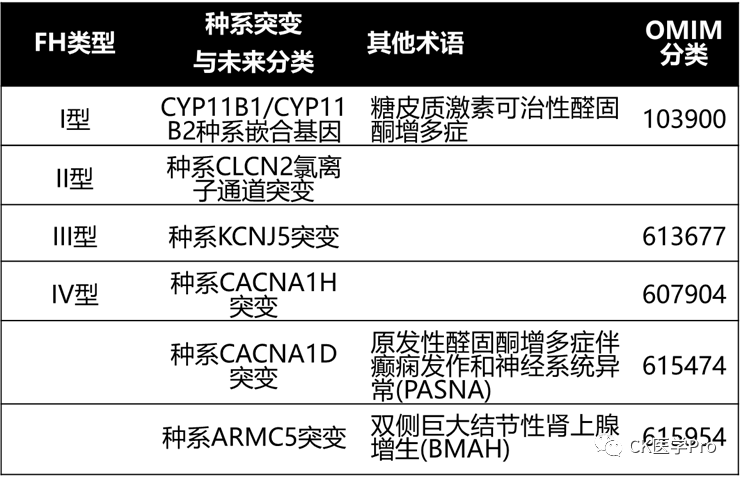
FH Type I: CYP11B1/CYP11B2 Germline Chimeric Gene
Glucocorticoid-remediable aldosteronism (GRA; FH Type I; OMIM #103900) is a form of aldosteronism that can be suppressed by physiological doses of glucocorticoids, thereby reversing excess aldosterone secretion[Ann Intern Med 1992; 116: 813–20]. FH Type I was first described in a single family in 1966[Can Med Assoc J 1966; 95: 1109–19]. 26 years later, the cause was discovered: a non-homologous exchange of the CYP11B1 gene (coding for 11β-hydroxylase) and the CYP11B2 gene (coding for aldosterone synthase) on chromosome 8q24.3, resulting in the CYP11B1/CYP11B2 chimeric gene, where the production of mineralocorticoids is regulated by adrenocorticotropic hormone rather than normal renin-angiotensin secretion[Nature 1992; 355: 262–5]. Continuous studies on 300 PA patients showed that the CYP11B1/CYP11B2 chimeric gene is rare; it was detected in only two patients, with a prevalence of 0.67%[Hypertension 2011; 58: 797–803]. The characteristics of the CYP11B1/CYP11B2 chimeric gene include early-onset hypertension, which can range from mild to severe. The diurnal characteristics of excess aldosterone in these patients may explain the rarity of hypokalemia. This chimeric enzyme is expressed throughout the adrenal cortex, leading to increased production of 18-hydroxycorticosterone and 18-hydroxycortisol due to C-18 hydroxylation and C-18 oxidation in the adrenal zona fasciculata. In the absence of glucocorticoid therapy, the CYP11B1/CYP11B2 chimeric gene leads to the excessive production of aldosterone, 18-hydroxycorticosterone, and 18-hydroxycortisol. Genetic testing for the CYP11B1/CYP11B2 chimeric gene should be considered in PA patients with a family history of PA, early onset (<20 years), or a family history of young strokes.
FH Type II: Germline Mutations of the CLCN2 Chloride Channel
FH Type II was first reported in 1991 and is inherited in an autosomal dominant manner[Clin Exp Pharmacol Physiol 1991; 18: 283–6; J Hypertens 2008; 26: 1577–82; Front Horm Res 2014; 43: 70–8; J Hum Hypertens 2011; 25: 560–4]. Families with FH Type II may have APA and IHA. FH Type II is more common than FH Type I but accounts for less than 6% of all PA patients. Linkage analysis studies showed association with the chromosomal region 7p22[J Hypertens 2008; 26: 1577–82; J Hum Hypertens 2011; 25: 560–4].
In a recent report on FH Type II families and 80 unresolved early-onset PA probands, the authors found germline CLCN2 chloride channel mutations in 8 of the probands[Nat Genet 2018; 50: 349–54]. All relatives of patients with early-onset PA carried the CLCN2 variants found in the probands. CLCN2 encodes a voltage-gated chloride channel expressed in adrenal glomerulosa cells. Mutated channels exhibit gain of function, opening at hyperpolarized membrane potentials, showing a higher probability of opening at the resting potential of the glomerulosa[Nat Genet 2018; 50: 349–54; Nat Genet 2018; 50: 355–61]. CLCN2 chloride channel mutations may be the cause of all or part of the patients classified as FH Type II. As mentioned above, FH Type II may prove to be a “placeholder” for a series of mutations that lead to PA(Table 15).
FH Type III: Germline Mutations in KCNJ5
FH Type III (OMIM#613677) was first discovered in a 5-year-old boy in 1959[AMA J Dis Child 1959; 98: 90–9], just four years after Conn initially described PA. In 2008, the familial nature of this disease became evident when two daughters of the proband/index case (aged 4 and 7) were diagnosed with severe PA[J Clin Endocrinol Metab 2008; 93: 3117–23]. Due to refractory hypertension, the father and both daughters underwent bilateral adrenalectomy during childhood. The adrenal glands showed significant hyperplasia. In 2011, pathogenic germline mutations were identified within this family: point mutations in KCNJ5, coding for the potassium channel kir 3.4 (gir 4), in or near the selective filter, were identified (Thr158Ala)[Science 2011; 331: 768–72]. This KCNJ5 mutation leads to a loss of ion selectivity, allowing sodium ions to enter, causing depolarization of glomerulosa cells; this is a signal for increased CYP11B2 expression, aldosterone production, and cell proliferation. To date, 12 families with six different KCNJ5 mutations have been identified[Proc Natl Acad Sci USA 2012; 109: 2533–8]:
-
p.Thr158Ala107;
-
p.Gly151Glu108-110;
-
p.Gly151Arg108,111,112;
-
p.Ile157Ser113;
-
p . Tyr152Cys114;
-
p.Glu145Gln110,115,116.
The estimated prevalence of germline KCNJ5 mutations is 0.3% among PA patients and 8% among familial PA patients[Hypertension 2012; 59: 235–40]. Most patients with germline KCNJ5 mutations present with polyuria, polydipsia, and refractory hypertension in childhood; surveys show both have significant hypokalemia and PA. In most cases, excess aldosterone secretion leads to the need for bilateral adrenalectomy. However, there is some heterogeneity in the age of presentation (oldest being 48 years); and some patients’ hypertension and hypokalemia can be controlled with MRA[Proc Natl Acad Sci USA 2012; 109: 2533–8; Hypertension 2012; 59: 235–40; Endocr J 2012; 59: 497–502; Horm Res Paediatr 2014; 82: 138–42]. Additionally, it has been reported that a patient with a germline KCNJ5 mutation also had adrenal-dependent Cushing’s syndrome[J Clin Endocrinol Metab 2016; 101: 4290–7].
FH Type IV: Germline Mutations in the CACNA1H Gene
FH Type IV (OMIM#617027) is inherited in an autosomal dominant manner, with incomplete penetrance, caused by mutations in the CACNA1H gene (located on chromosome 16p13) that encodes the α subunit of the L-type voltage-gated calcium channel (CAV 3.2)[Elife 2015; 4: e06315; EBioMedicine 2016; 13: 225–36]. Among 40 unlinked patients affected by childhood hypertension and PA, five were found to have a new germline CACNA1H mutation (p.Met1549Val)[117]. Four additional germline CACNA1H mutations (p.Met1549Ile, p.Ser196Leu, p.Pro2083Leu, and p.Val1951Glu) have also been reported in PA patients[EBioMedicine 2016; 13: 225–36].
Commercial testing for germline CACNA1H mutations is available and should be considered in children with aldosteronism and in families with more than one PA member. Treatment for patients with germline CACNA1H mutations should be the same as for those with significant sporadic PA caused by IHA. However, in specific cases of refractory disease associated with bilateral adrenal massive hyperplasia, laparoscopic bilateral adrenalectomy may need to be considered.
PA with Seizures and Neurological Abnormalities: Germline Mutations in CACNA1D
PA with seizures and neurological abnormalities (PASNA) is caused by de novo germline mutations in CACNA1D (OMIM#615474). CACNA1D encodes the α1D subunit of the L-type voltage-gated calcium channel (Cav 1.3), located on chromosome 3p14.3. PASNA has been reported in two children with PA, seizures, and neurological abnormalities. Severe neurological abnormalities have prevented these individuals from reproducing; thus, despite being germline mutations, they do not form a familial form of PA. Additionally, a missense germline mutation in CACNA1D (p.Val104Leu) has been reported in a patient with autism and seizures[Hum Mol Genet 2017; 26: 2923–32]. The clinical phenotype associated with germline CACNA1D mutations is likely to be proven as a variant of PASNA.
PA and ARMC5 Mutations
In patients with bilateral large nodular hyperplasia, heterozygous germline mutations in ARMC5 are most commonly associated with subclinical glucocorticoid secretion autonomy or Cushing’s syndrome[Endocr Relat Cancer 2018; 25: R131–52; N Engl J Med 2013; 369: 2105–14]. However, it has been reported that patients with PA and Cushing’s syndrome have ARMC5 mutations due to bilateral large nodular hyperplasia, which is most common in African American patients[J Hum Hypertens 2016; 30: 374–8; PLoS ONE 2018; 13: e0191602].
Somatic Driver Mutations
Additionally, regarding somatic driver mutations, it is currently believed that somatic driver mutations in the KCNJ5, ATP1A1, ATP2B3, CACNA1D, and CTNNB1 genes significantly participate in the pathogenesis of primary aldosteronism. It is speculated that nearly 100% of APAs will ultimately be associated with somatic driver mutations; details can be found in the previous section on mechanisms.
Cortisol/Aldosterone Co-secretion
Is this a clinical subtype that is not classic? This is a clinical situation that needs attention.
Patients with PA exhibit an increased risk of insulin resistance and metabolic syndrome, depression, and osteoporotic fractures. These associations seem to align with autonomous or excessive secretion of glucocorticoids. A steroid metabolomics study compared the following patient groups: 174 PA patients (103 APA, 71 IHA); 162 healthy controls; 56 endocrine-inactive adrenal adenomas; 104 patients with mild autonomous glucocorticoid secretion; and 47 patients with adrenal-dependent Cushing’s syndrome[JCI Insight.2017;2(8)]. The main findings were as follows:
-
Compared to the control group and patients with subclinical Cushing’s syndrome, patients with PA (APA and IHA) had significantly increased excretion of cortisol and total glucocorticoid metabolites (p < 0.001);
-
Metabolic risk surrogate parameters associated with glucocorticoids but not related to aldosterone excretion;
-
In APA patients, unilateral adrenalectomy resolved both mineralocorticoid and glucocorticoid excess.
These findings indicate that some degree of cortisol co-secretion is very common among PA patients and is associated with metabolic risk, which may suggest that the use of MRA in IHA and APA patients may not prevent glucocorticoid-dependent metabolic risk. However, these data should be interpreted with caution, as this study is not only about determining the cortisol co-secretion in APA but also the increased steroid metabolomics in APA and IHA patients. The hypothalamic-pituitary-adrenal axis in these patients is not suppressed, and APA patients receiving surgical treatment did not experience adrenal crises or steroid withdrawal.
APA can co-secrete cortisol, which may have clinical implications for perioperative management. Subclinical Cushing’s or relatively obvious autonomous cortisol secretion + APA co-secretion tumors may require glucocorticoid replacement after resection to avoid the risk of adrenal insufficiency.
Additionally, co-secretion may produce significant biases when performing AVS, such as when using cortisol for data correction.
When should clinicians test for cortisol co-secretion in APA patients? Generally, clinically significant cortisol secretion from adrenal adenomas is related to tumor size. Unlike aldosterone secretion from adenomas, clinically significant cortisol secretion requires a “big factory” (usually adenomas larger than 2 cm). Therefore, it is reasonable to test for cortisol secretion in PA patients when the adrenal adenoma diameter exceeds 1.5 cm. Such tests include baseline dehydroepiandrosterone sulfate testing and overnight 1-mg dexamethasone suppression testing.
When autonomous glucocorticoid secretion is observed in PA patients with a single cortical adenoma larger than 1.5 cm, AVS may be considered unnecessary.
Clinicians do not have good long-term drug treatment options for Cushing’s syndrome, while MRA can effectively treat PA. In rare cases of PA with Cushing’s syndrome, there may be independent adenomas secreting aldosterone and cortisol simultaneously.
Endocrine Metabolic Disease @CK Medicine
Endocrine Metabolic Disease Knowledge Framework @CK Medicine
Endocrine Metabolic Disease Graded Diagnosis and Treatment @CK Medicine
PS: Endocrinologists who want to join the professional group can add WeChat CK-ENDO; only for endocrinologists; please indicate identity when adding WeChat: XX Hospital – Department – Name – Title, otherwise, the group will be refused. After joining, please also change your group nickname to: XX Hospital – Department – Name. The professional group is limited to communication and learning among endocrinologists and is currently not open to personnel from other departments or patients.In fact, those who can endure such dry specialty content and see this “PS” are basically only endocrinologists left , but if you are a non-endocrinologist and are so interested in this content that you have read these two paragraphs of PS, and even want to join the group, then just follow the steps in the PS, welcome.
, but if you are a non-endocrinologist and are so interested in this content that you have read these two paragraphs of PS, and even want to join the group, then just follow the steps in the PS, welcome. By reviewing the latest guidelines, consensus, and review literature, on one hand, clinical practice can become more solid, avoiding arbitrary judgments and following, while practicing based on a solid evidence base; on the other hand, under the framework of standardized clinical practice, new and more scientific clinical issues can be raised, tracked, and resolved. During the literature review process, I also continuously discover and raise questions based on my knowledge background and clinical practice, but due to the limitations of clinical models, I may not be able to immediately design solutions to problems, but I believe that many things are “what you think of will eventually resonate.” Moreover, I am pleased that some doctors have been able to incorporate the content of new developments from this public account into clinical practice to discover and attempt to solve clinical problems, or to refine new ideas and concepts in clinical design. If you are willing to communicate or collaborate, please leave a message in the background.
By reviewing the latest guidelines, consensus, and review literature, on one hand, clinical practice can become more solid, avoiding arbitrary judgments and following, while practicing based on a solid evidence base; on the other hand, under the framework of standardized clinical practice, new and more scientific clinical issues can be raised, tracked, and resolved. During the literature review process, I also continuously discover and raise questions based on my knowledge background and clinical practice, but due to the limitations of clinical models, I may not be able to immediately design solutions to problems, but I believe that many things are “what you think of will eventually resonate.” Moreover, I am pleased that some doctors have been able to incorporate the content of new developments from this public account into clinical practice to discover and attempt to solve clinical problems, or to refine new ideas and concepts in clinical design. If you are willing to communicate or collaborate, please leave a message in the background.