Sharing the essence of recent articles from the “China Medical Device Information” magazine, with continuous updates! Welcome to subscribe! Welcome to contribute! Follow our WeChat public account:zgylqxxx
Source: This article was published in the “China Medical Device Information” magazine, 2022, Vol. 28, No. 9
Author: He Ting Bai Ru*
Affiliation: Shanghai Medical Device and Cosmetic Review Center (Shanghai 200020)
Abstract: In the quality management of the entire lifecycle of in vitro diagnostic (IVD) reagents, finished product testing is an important quality assurance measure before the product is marketed, while sampling testing is a requirement for continuous monitoring of product quality based on the characteristics of such products after they are on the market. This article summarizes the specific requirements for finished product testing and sampling testing of IVD reagent products based on current regulatory requirements, aiming to provide a reference for the formulation and implementation of relevant work specifications for IVD reagent production enterprises.
Keywords: In Vitro Diagnostic (IVD) Reagents, Finished Product Testing, Sampling Testing
Quality control is the set of operational techniques and activities aimed at achieving quality requirements. By monitoring the quality formation process, it eliminates factors that cause non-conformities or unsatisfactory results at all stages of the quality chain. Its goal is to ensure that the quality of products or services meets the requirements (including explicit, implicitly understood, or mandatory regulations) to meet quality standards and achieve economic benefits. For in vitro diagnostic (IVD) reagent products, this includes the preparation of small samples and performance validation based on market demand and clinical needs during the design and development phase of new products; batch sample pilot production during the registration phase; and the final finished product, which includes testing according to established performance indicators and quality reviews after the product is on the market, all of which affect the safety and effectiveness of the product after it is marketed. The specific requirements for people, machines, materials, methods, and environment in these processes constitute the basic chain of quality control for this product.
How can we ensure the quality of IVD reagent products awaiting market release? How can we continuously and traceably monitor the quality of products after they are on the market? To address these issues, enterprises must control different stages of the product lifecycle. Among these, finished product testing and sampling testing are essential components of the entire quality control chain. Failures in these process controls can directly affect the performance indicators of marketed products, impact the accuracy and reliability of diagnostic results, and subsequently affect the quality of clinical auxiliary diagnosis, as well as have significant implications for precise diagnosis and treatment in medical institutions, patient safety, and rational medication/treatment.
1. Regulatory Requirements for Finished Product Testing and Sampling Testing of IVD Reagents
The newly revised “Regulations on the Supervision and Administration of Medical Devices” (State Council Order No. 739) [1] has been implemented since June 1, 2021. The new regulations further emphasize the requirements for quality management, stating: “Article 30: Those engaged in the production of medical devices should possess: (2) institutions or dedicated inspection personnel and inspection equipment capable of conducting quality inspections of the produced medical devices”; “Article 33: The quality management specifications for medical device production should clearly define matters affecting the safety and effectiveness of medical devices, including design and development, production equipment conditions, raw material procurement, production process control, product release, organizational structure, and personnel allocation”; “Article 35: Medical device registrants, filers, and entrusted manufacturers should ensure that the medical devices they produce meet mandatory standards and the technical requirements of registered or filed products”; and the first clause of Article 86 regarding penalties states, “Producing, operating, or using medical devices that do not meet mandatory standards or do not comply with the technical requirements of registered or filed products.” Article 7 of the “Measures for the Supervision and Administration of Medical Device Production” (Order No. 7 of the National Medical Products Administration) [2] also clearly states that “Those engaged in medical device production should meet the following conditions: (2) have institutions or dedicated inspection personnel and inspection equipment capable of conducting quality inspections of the produced medical devices.”In China, in accordance with the management of medical devices, in vitro diagnostic reagents refer to products used for the in vitro detection of human samples in the processes of disease prediction, prevention, diagnosis, treatment monitoring, prognosis observation, and health status evaluation, which can be used alone or in combination with instruments, apparatus, devices, or systems. As an important component of domestic medical devices, relevant regulatory documents have made clear requirements for the quality control of IVD reagent products.In addition to the aforementioned general regulatory documents, at the implementation level, the National Medical Products Administration has issued the “Registration Management Measures for In Vitro Diagnostic Reagents” (Order No. 5 of the National Medical Products Administration) [3]; “Appendix for Quality Management Specifications for Medical Device Production: In Vitro Diagnostic Reagents” [4]; and “Guidelines for On-Site Inspection of Quality Management Specifications for Medical Device Production: In Vitro Diagnostic Reagents” [5]. To guide medical device production enterprises in effective quality management according to the technical requirements of registered or filed products, the administration has organized the formulation and release of the “Guidelines for Quality Control and Product Release for Medical Device Manufacturing Enterprises” [6].In summary, the quality stability of an IVD product awaiting market release needs to be comprehensively controlled and ensured at all stages of the quality chain, from the initial design of the product, sample stability pilot production, process inspection, finished product testing, and stability assessment of samples after the product is on the market, with finished product testing and stability assessment of samples constituting key control points in the quality control of the product lifecycle.
2. Key Control Points of Finished Product Testing
Finished product testing refers to the comprehensive inspection and testing of completed products to ensure that the performance of finished products meets relevant requirements and guarantees their effectiveness. Finished product testing is a key step in quality control for IVD reagent products before they are marketed. Enterprises should control aspects such as personnel responsibilities and training; installation, use, and maintenance of equipment; and the output of normative documents and traceability of records. Specifically, the key control contents of finished product testing can be summarized as follows.First, enterprises should be equipped with appropriate human resources, especially focusing on the specific operators of finished product testing and personnel responsible for product release before marketing. Responsibilities and authorities of the quality management department and relevant personnel should be clearly defined, and finished product testing operations should be completed by appointed personnel with relevant professional knowledge and practical experience who have undergone job training. Enterprises should pay attention to the capabilities of the quality responsible person and product release personnel.Second, enterprises should set up appropriate inspection areas based on specific product characteristics and production scale, and equip them with inspection equipment that meets the requirements of finished product testing, regularly verifying the capacity of this equipment and managing it daily. For equipment requiring measurement, a regular measurement plan should be established to ensure compliance with measurement audits. For finished product inspections involving multiple devices, the impact of differences between devices on test results should be considered. To ensure the traceability of quality system operations, records of the main equipment’s usage and maintenance should be retained to guarantee the applicability of the equipment.Third, enterprises should develop finished product testing procedures based on mandatory standards and the technical requirements of registered or filed products, delivery requirements, and internal control standards. Finished product testing procedures should determine the verification/confirmation/monitoring/measuring/testing procedures and requirements that need to be routinely controlled for finished products, ensuring that each batch of finished products meets acceptance criteria. Records of acceptance and rejection and related disposal measures should be kept [6].The finished product testing procedure should at least specify the name, specifications, verification/confirmation/monitoring/measuring/testing items and methods, applicable instruments and tools, sampling procedures, sampling plans, acceptance criteria, referenced standards/referenced measurement procedures, and related records. The sampling plan should have a statistical basis and analyze the confidence level of statistical inference to ensure that the sampled samples are representative [6].In principle, the content of the finished product testing procedure should cover the inspection items and methods that need routine control in the registered or filed product technical requirements. If there are items that cannot be covered, they should be explained in the finished product testing procedure. If necessary, confirmed alternative solutions should be provided [6].Overall, enterprises should ensure that the finished product testing procedures and specific operational guidelines are consistent with regulatory approvals; the operability of operational guidelines and the rationality of established testing rules.Fourth, the final finished product testing report should meet basic traceability requirements, and the report should at least include information on the samples used for testing, supporting reagents and consumables information, equipment information, and operators. Each performance indicator’s detection should retain original inspection records, corresponding to the items and results in the inspection report.Fifth, enterprises should establish and implement calibration material traceability procedure documents based on product technical requirements and regulatory approvals, paying attention to the potential impact on products caused by using different batches of higher-level reference materials, establishing internal control deviation acceptance standards, and retaining evaluation reports and original records where applicable.Sixth, enterprises should establish and implement finished product release procedures, clarifying conditions for product release and approval requirements. Before release, finished products should at least meet the following conditions: completion of all prescribed processes; completeness of specified batch production records; completeness of all specified quality control records regarding incoming materials, processes, finished product inspections, verifications, etc., with results meeting specified requirements, and records issued by testing/verification personnel and their review and approval personnel according to regulations [6].
3. Requirements for Sampling Testing
Product sampling refers to the samples preserved by manufacturing enterprises according to regulations, used for quality traceability or investigation, as well as product performance research. Regular performance testing and quality stability retrospective evaluation of such samples help identify issues during quality traceability of medical device products, investigation of adverse events, clarify accident responsibilities, and provide data support for confirming or modifying product technical indicators.First, enterprises should establish sampling management systems to clarify the purpose of sampling, sampling samples, sampling observation requirements, and sampling ratios/quantities. For IVD reagent products, the purposes of sampling are generally divided into three categories: for product quality traceability; for raw material quality traceability; and for stability studies.The samples used for sampling should meet the following basic requirements: the samples must be products that have passed inspection; they should be randomly selected from the finished product batch, having completed all production processes and capable of representing the sampled batch; the packaging form should be consistent with the form for market sale; and the model specifications of the sampled products should be clearly defined.The sampling area for storing samples should meet the following basic requirements: the sampling area should be relatively independent and sufficient to store samples; in principle, the storage conditions should be consistent with those of finished products; the sampling area should be equipped with environmental monitoring equipment that meets product quality characteristics requirements, regularly monitored, and relevant records retained.The sampling ratios/quantities should be determined by the enterprises themselves but should at least support one quality traceable inspection. For special purposes of sampling, such as new process research or extension of validity periods, separate sampling should be conducted without affecting quality traceable inspection.Enterprises should clarify the frequency/cycle of sampling testing/sampling observation based on different sampling purposes. Generally speaking, the observation time for sampling should not be less than the product validity period. For stability study samples, the observation time may be appropriately extended or the observation frequency increased. Enterprises should clarify the items, testing methods, and judgment criteria for sampling testing/sampling observation and possess corresponding testing capabilities.The sampling testing/sampling observation process should generate corresponding records, including at least: sampling ledgers, sampling testing/sampling observation records, and reports. The inspection/observation records and reports should specify the sampling batch number, inspection/observation date, inspection/observation personnel, and inspection/observation results.Upon expiration of the sampling period, a summary, analysis, and archiving of the sampling testing/sampling observation reports should be conducted to evaluate product quality and examine the stability of product quality. If non-conformities are found during the inspection/observation process, they should be handled according to relevant regulations and the causes of non-conformity analyzed. For remaining samples after the sampling period, enterprises should handle them according to relevant regulations to prevent unexpected use of sampling samples.
4. Common Issues
Based on the summary data of daily on-site system inspections, the “Guidelines for On-Site Inspection of Quality Management Specifications for Medical Device Production: In Vitro Diagnostic Reagents” [5] consists of eleven chapters, among which non-conformities related to “Chapter 8: Quality Control” have accounted for a high proportion in daily system inspections (see Figure 1).
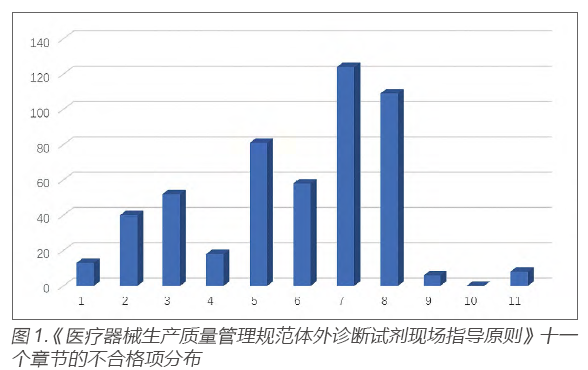
Regarding the specific provisions of Chapter 8, the distribution of non-conformities also shows significant differences, mainly concentrated in clauses *8.3.1, 8.7.1, 8.10.1, etc. (see Figure 2).
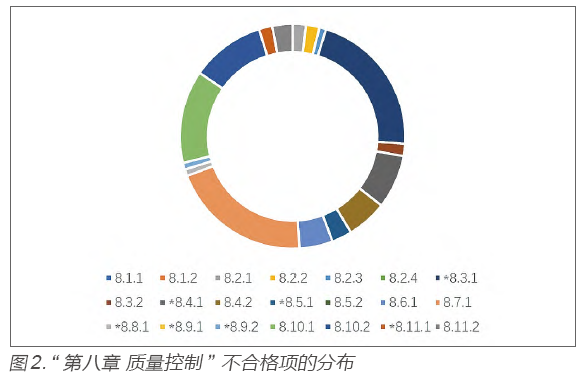
In summary, common issues faced by IVD reagent production enterprises can be roughly summarized as follows.First, the rationality of normative document formulation and the compliance of actual operations, such as: the inspection indicators in the finished product release inspection procedures do not cover the approved product technical requirements or the testing methods are below the product technical requirements, and no assessment or control of these discrepancies has been made; no inspection rules for finished product release or sampling testing have been established; the sampling testing procedures do not clearly state the quantity, inspection cycle, and inspection content for different purposes of sampling; no operational guidelines for the preparation and valuation of enterprise reference materials have been established; and actual release processes before product marketing do not align with release-related regulations.Second, issues with traceability of specific inspection records, such as: “accuracy” testing batch numbers of enterprise reference materials not being recorded in inspection reports; missing information on high-concentration/low-concentration serum samples used for “linear range” testing; incomplete information on serum samples used for “within-batch precision” testing (including batch numbers, indicated values, etc.); incomplete ledger information for reference materials (especially self-made internal calibration materials) (such as lack of key technical indicators, traceability paths, etc.); and incomplete information on human-derived sample lists used for preparing testing samples (such as original sample numbers, clinical information, etc.).Third, insufficient validation work, such as: most international standards or national standards supplied for procurement do not specify specific validity periods, and enterprises generally define their own expiry dates, but some enterprises lack corresponding validation data support, such as: no established cycle for revalidation of relevant reference materials; enterprises defining the validity period of primary reference materials as 2 years but failing to provide validation documentation for the validity period.
5. Conclusion
With the rapid development of domestic biopharmaceutical technology platforms, the entire domestic diagnostic reagent industry is continuously achieving technological breakthroughs and innovations, gradually breaking the monopoly position of imported reagents and diagnostic instruments. The reliability and stability of domestic IVD reagent products are fundamental conditions for expanding broad market prospects. Enterprises should establish reasonable requirements for finished product testing and sampling testing based on product characteristics, methodological platforms, clinical usage needs, and regulatory requirements to ensure the effectiveness and safety of marketed products, and conduct regular retrospective evaluations and analyses of quality.
References Omitted
Review of Previous Content
[Academic Sharing] Current Status and Trends in the Development of Domestic and International In Vitro Diagnostic Reagent Industry
[Academic Sharing] Overview of the Current Development Status and Future Trends of the Medical Robotics Industry Based on Data Analysis
[Academic Sharing] Key Points of Clean Area Air Supply and Exhaust Systems in Medical Device Production and Their Inspection
[Academic Sharing] Discussion on the Latest Adjustments in FDA Approval Requirements for High-Risk Medical Devices
[Academic Sharing] Common Issues and Analysis of the Quality Management Specifications for Medical Device Production
[Academic Sharing] Research on Clinical Evaluation Methods for Class II Medical Devices
About This Journal
“China Medical Device Information” magazine is a national-level official publication registered with the National Press and Publication Administration, issued nationwide, and is the only comprehensive medical device technology magazine supervised by the National Medical Products Administration. The magazine was founded in 1995 and is a “statistical source journal” of the “China Academic Journal Comprehensive Evaluation Database (CAJCED)”, a “full-text inclusion journal” of the “China Journal Full-text Database (CJFD)”, a “core journal” of the “Chinese Core Journals (Selection) Database”, a “Chinese Scientific and Technological Journals Database” inclusion journal, and a “Chinese Biomedical Journals Database (CMCC)” inclusion journal.
“China Medical Device Information” magazine is a semi-monthly publication of large sixteen open (210mm×285mm), using imported high-quality coated paper, full-color printing. The publication dates are the 15th and 25th of each month. “China Medical Device Information” has complete qualifications for publication, has postal delivery qualifications (postal delivery code 82-256), and the distribution methods include: postal subscriptions, self-operated subscriptions, complimentary distribution, and conference distribution. The average distribution volume is 50,000 copies per issue, and there will be an increase in distribution during large exhibitions and academic events.
We sincerely welcome industry experts, scholars, and users of medical equipment, as well as R&D designers to contribute to this journal and work together to make this publication better.
Submission Guidelines
Submission Email: [email protected] [email protected]