[Multiwfn+VMD] Plotting Crystal Hirshfeld Surface and Fingerprint Diagram – 2
![[Multiwfn+VMD] Plotting Crystal Hirshfeld Surface and Fingerprint Diagram - 2](https://boardor.com/wp-content/uploads/2025/10/92bf5286-0bc0-43e7-8630-7e20cf4f1961.jpg)
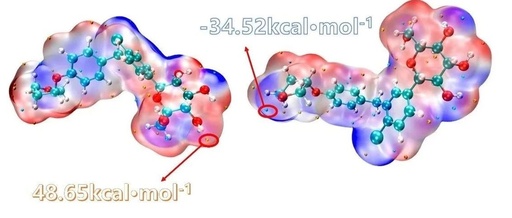
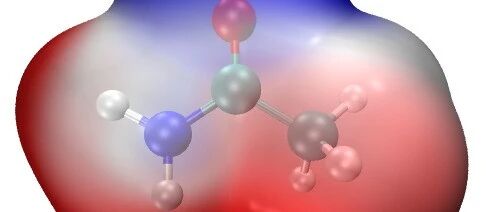
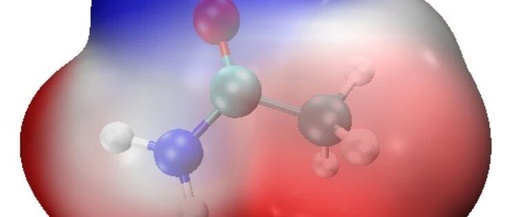
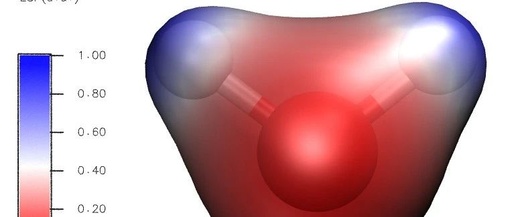
This article is a supplement to the content of [Multiwfn+VMD] Plotting Crystal Hirshfeld Surface and Fingerprint Diagram. A preliminary analysis of intermolecular interactions has been completed, but how to specifically distinguish each type of interaction remains unclear. This article can be ignored if the study does not require the separation of these weak interactions. For … Read more