The Technion – Israel Institute of Technology Professor Ilan Marek’s team utilized α-boryl carbanion intermediates as a platform for stereochemical generation, exploring the complex behavior of α-boryl carbanions generated from various cyclopropyl boronic methyl iodides in selective ring-opening and electrophilic reactions, conducting a series of highly stereoselective nucleophilic substitution reactions to construct vicinal stereocenters.
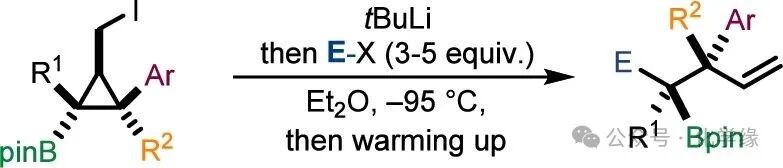
Abstract
We report the generation and stereocontrolled electrophilic trapping of α-boryl carbanions via the selective anionic ring-opening of stereodefined iodomethyl cyclopropylboronic esters. The strategy relies on a lithium–iodine exchange to generate borata alkene intermediates, affording vicinal tri- and tetrasubstituted boronic esters with excellent levels of diastereoselectivity after electrophilic trapping. The stereocontrol arises from conformational preferences of the boron alkylidene, driven by its unique electronic structure. This work expands the synthetic utility of α-boryl carbanions and demonstrates their potential in the stereocontrolled construction of sp3-rich organoboron frameworks.
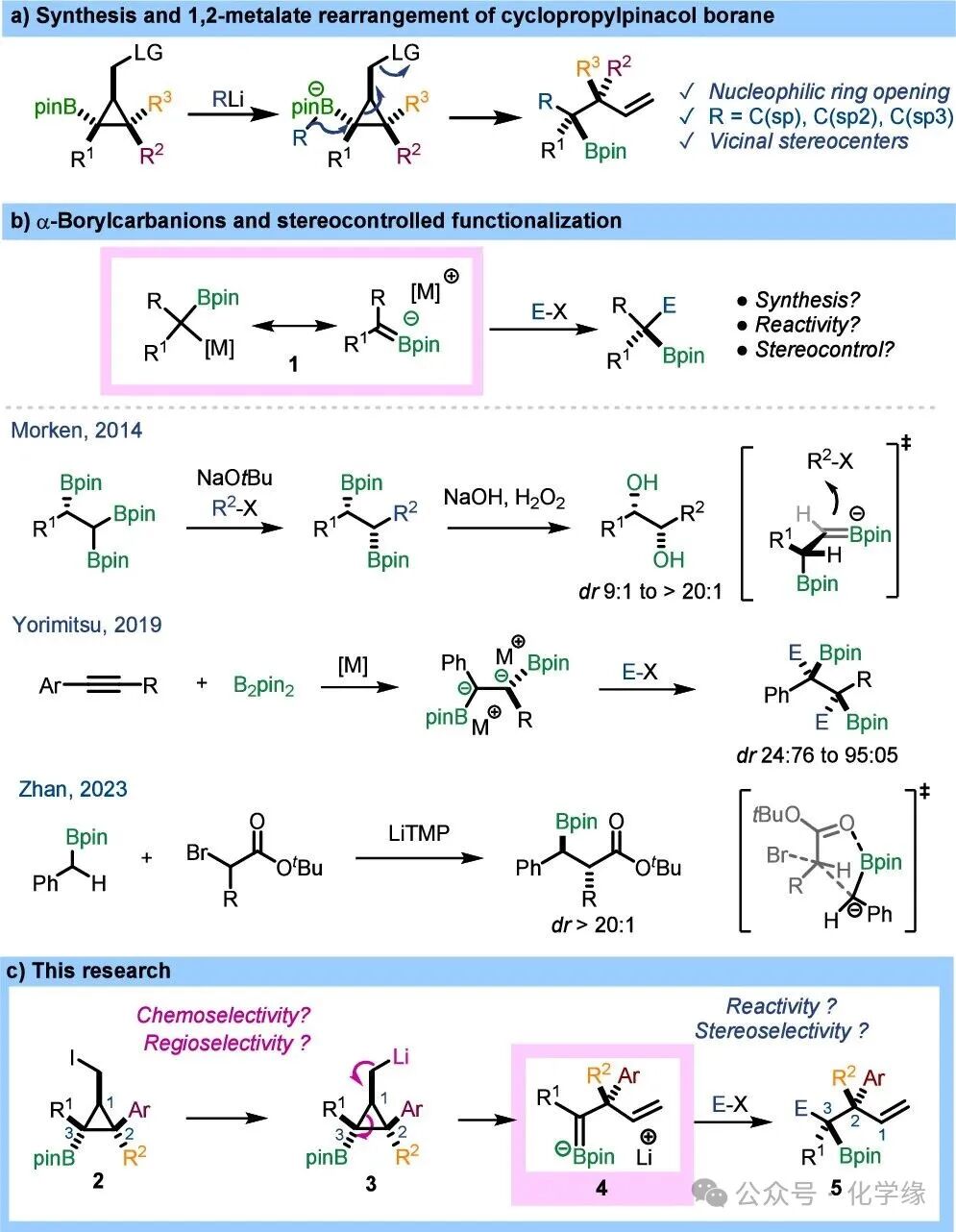
Figure 1 Current research status and possible transformations
Organoboron compounds have become highly valuable synthetic intermediates due to their multifunctional functionalization reactions (such as Suzuki–Miyaura cross-coupling, oxidation reactions, Matteson homologation reactions, metal-catalyzed enantioselective reactions, and transformations into various stereochemical configurations of organometallic species). Given the widespread applications of organoboron compounds, synthesizing boron-rich compounds with adjacent chiral centers remains strategically significant. Particularly attractive is the construction of vicinal tri- and tetrasubstituted chiral centers containing boronic esters in non-cyclic systems. Previous reports have synthesized non-cyclic boronic esters with tertiary and quaternary centers through the ring-opening of stereodefined cyclopropyl boronic esters. This method relies on adding organolithium reagents to cyclopropyl boronic esters containing suitable leaving groups, forming boronic ester complexes. Subsequently, a 1,2-metal rearrangement occurs accompanied by a ring-opening reaction, generating tertiary boranes with excellent diastereoselectivity to achieve the desired adjacent chiral centers. Inspired by this work and recognizing the importance of these stereochemical molecular frameworks, we considered whether they could be obtained through a distinctly different approach—namely, the non-diastereoselective reaction of α-boryl carbanions with electrophiles.
α-Boryl carbanion1 (also known as borylalkene or boronic alkene) is stabilized by electron delocalization towards the boron atom, forming a πC–B bond. In recent years, such intermediates have become multifunctional synthetic intermediates capable of various transformations: including cyclization and ring-opening reactions, additions to aldehydes and imines, conjugate addition reactions, allylation reactions, and net [2+2] cycloaddition reactions.
Despite these significant advances, the intermolecular nucleophilic substitution reactions with isomeric selectivity still face substantial challenges. Morken reported that the deborylation of 1,1,2-tris(boronate) can generate α-boryl carbanions, which can yield 1,2-diols with high yields and excellent isomeric selectivity after alkylation and subsequent oxidation reactions. This selectivity is attributed to the interaction between the σC–B orbitals and the adjacent πC–B orbitals, which is believed to stabilize specific conformations of the carbanion, thereby guiding the selective capture of electrophiles. Yorimitsu observed varying degrees of isomeric selectivity when generating non-cyclic products from dianions formed by the reduction of diboronic acids substituted with aryl acetylene. Recently, Zhan reported the stereoselective deprotonation of benzyl boronic esters, which was rationalized through a five-membered ring chelation model. The authors proposed the preparation of boronic alkenes4 from the stereochemical cyclopropyl boronic ester2. This sequence begins with a lithium–iodine exchange reaction, facilitating the selective ring-opening of the resulting lithium cyclopropyl boronic ester3 to obtain the desired intermediate boronic alkene4. This intermediate can then be captured by electrophiles to generate ring-opened boronic esters containing adjacent chiral centers5. This strategy is expected to overcome the synthetic bottleneck of highly substituted boronic ester compounds and can directly yield boronic alkene without the need for additional stabilizing groups. However, to realize this strategy, the following key challenges must be addressed:
1. How to achieve the target lithium–iodine exchange while avoiding the formation of 1,2-metal acid salt rearrangement of the tertiary boronic ester salt complex?
2. How to guide the ring-opening reaction of compound3 to selectively cleave only the C1–C3 bond rather than the C1–C2 bond? Especially considering that the latter would generate a benzyl carbanion?
3. How to control the isomeric selectivity in the reaction with electrophiles to obtain a single isomer of compound5?
Figure 2 Synthesis of cyclopropyl boronic methyl iodides
To address these issues, a series of cyclopropyl methyl iodides2 were synthesized using previously developed methods. This strategy is based on the copper-catalyzed hydroxylation and carbon-boration reactions of cyclopropyl esters. The resulting esters were reduced to the corresponding alcohols, which were then converted to mesylates, and finally obtained the target iodides through Finkelstein exchange reactions. This modular process allows for a wide range of substitution patterns. A series of cyclopropyl boronic methyl iodides2a-n with various aryl substituents (including electron-rich, electron-poor, and heteroaryl substituents) were successfully synthesized, achieving excellent control in regioselectivity and enantioselectivity.
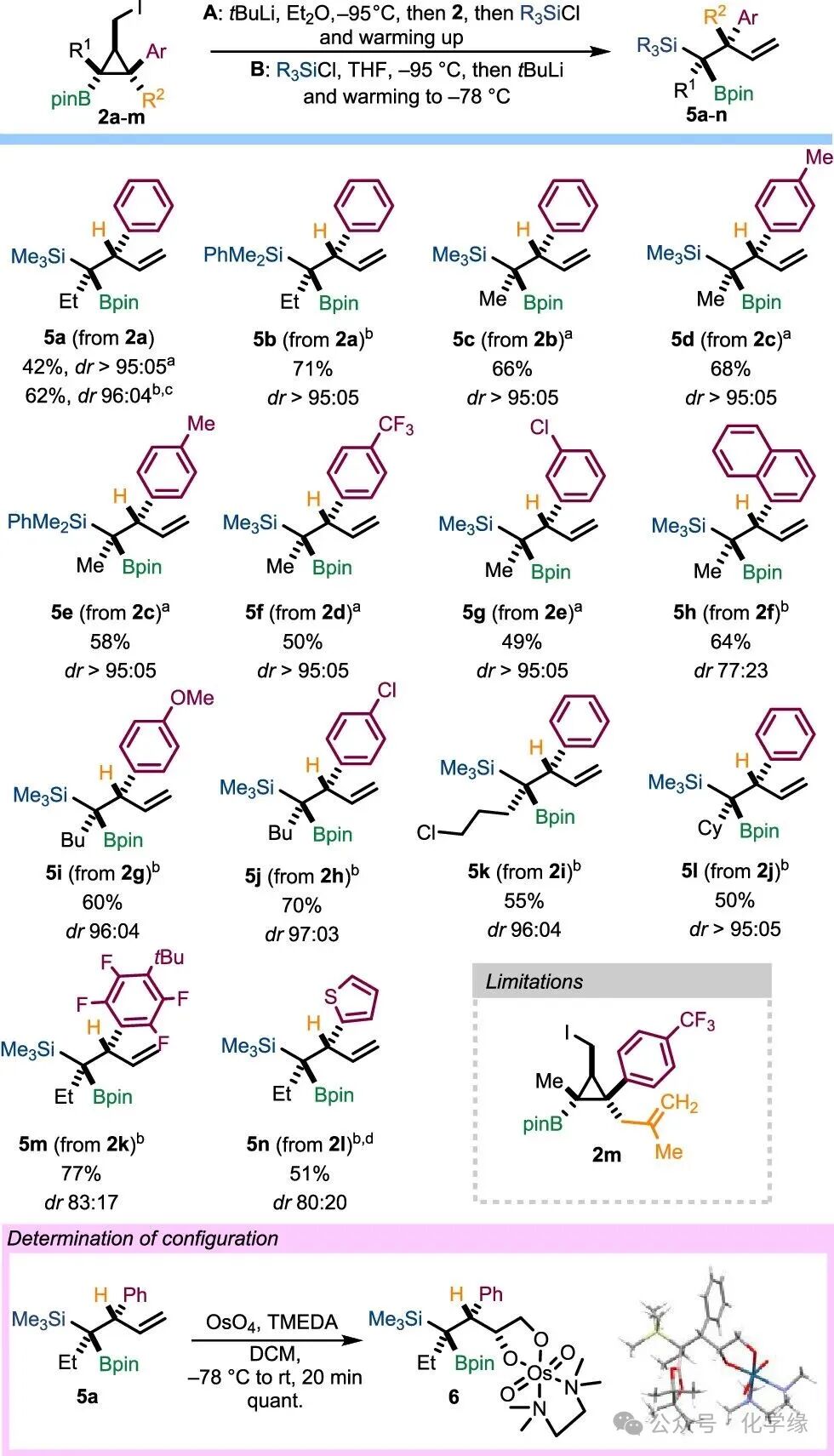
Figure 3 Scope of ring-opening/silylation reactions
By obtaining various cyclopropyl boronic methyl iodides2, the authors investigated the complex behavior of the resulting α-boryl carbanions in selective ring-opening and electrophilic reactions. After comprehensive optimization of the reaction conditions, the optimal conditions for generating α-boryl carbanion4 were determined: using tert-butyllithium in diethyl ether at -95 °C for lithium–iodine exchange, followed by the addition of Me3SiCl (Condition A). Under these conditions,5a was obtained in moderate yield with excellent enantiomeric ratio. In an alternative approach, if Me3SiCl is added to the reaction system beforehand, the reaction can be conducted in THF (instead of diethyl ether) (Condition B).
Further studies explored the application range and limitations of this reaction. Both sets of reaction conditions produced the same stereochemical results, but there were slight differences in yield and enantiomeric ratios. When2a was subjected to simultaneous ring-opening and silylation reactions, whether using Me₃SiCl or PhMe₂SiCl as electrophiles, efficient conversion occurred, yielding the target products5a and5b with excellent selectivity. Similarly, clean conversions were observed in the series of substrates2b-e, regardless of whether they contained electron-rich or electron-poor aryl groups, yielding single enantiomers of boronic esters5c–g. The naphthyl-substituted tertiary borane5h was also synthesized with a yield of 66%, but with moderate enantiomeric ratio. Extending the alkyl chain on R1 to butyl and cyclohexyl did not affect the reaction results, with the corresponding products5i-l obtained with yields of 50–70% and good selectivity. Notably, compounds containing functional groups within the R1 chain2i exhibited good tolerance under the reaction conditions, yielding silylated chlorinated alkyl compounds5k with a yield of 55%.
Further limitations of this method were revealed. For example, the ring-opening reaction of perfluorophenyl compound2k proceeded with a high yield of 77% and moderate enantiomeric ratio, generating a single product5m through simultaneous ring-opening, silylation, and t-BuLi-mediated ortho-selective SNAr reactions. For the trans-configured phenyl bromide and methylene iodide2l, the reaction also yielded alkyl silanes with the same stereochemical results, indicating that the stereochemistry at C1 does not affect the reactivity. A slight over-silylation phenomenon was observed, possibly due to the neighboring lithiation of the thiophene ring being captured by Me₃SiCl. Compound2m generated a complex mixture, presumably due to its high steric hindrance and the sensitivity of the alkene functional group. When5a was prepared on a 700 mg scale, both yield and enantiomeric ratio did not significantly decrease.
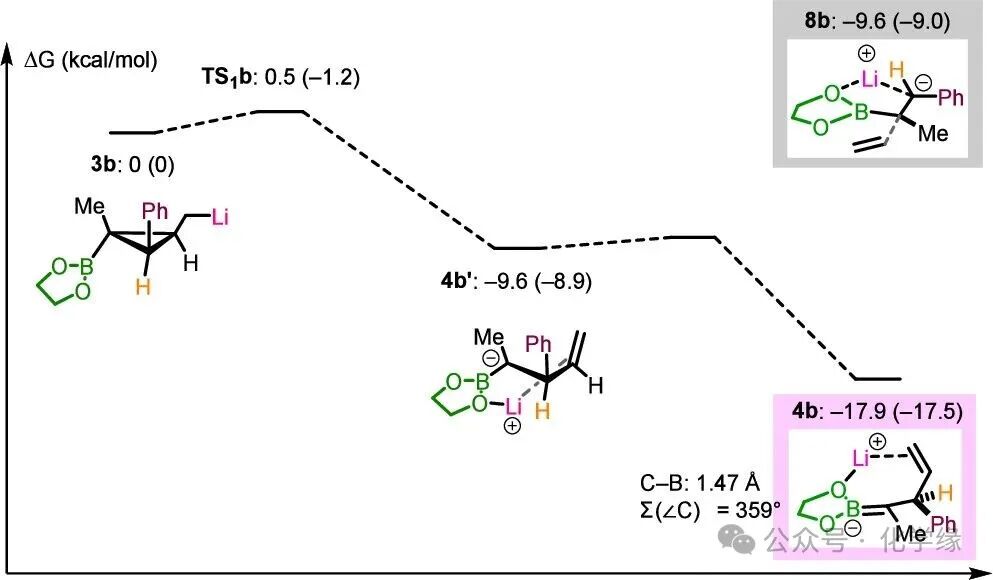
Figure 4 Ring-opening reaction of lithium cyclopropyl boronic esters
Given the observed high enantiomeric selectivity, density functional theory (DFT) calculations were employed to investigate the mechanism of this reaction. Substrate2b was selected as a representative substrate, and Me₃SiCl was used as a model electrophile. To simplify the system, we modified the boronic ester to an ethylene glycol derivative.
First, the ring-opening reaction of cyclopropyl methyl lithium3b was studied, which generates the α-boryl carbanion intermediate4b. This process begins with the formation of a transient intermediate4b′, which occurs through a nearly barrierless transition state (TS1b,ΔΔG⧧=0.5 kcal/mol). Subsequently, the intermediate4b′ undergoes a near-zero barrier isomerization reaction to form a more stable intermediate4b. This intermediate features borylalkene characteristics: a shorterC-B bond length (1.47 Å, while3b is1.54 Å) and a planar carbanion center (∑(∠C) = 359°, while3b is351°). The possibility of C1–C2 bond cleavage generating a benzyl anion8b was examined. It was found that the thermodynamic entropy of8b is higher than that of4b by 8.3 kcal/mol, indicating that thermodynamics favors the observed ring-opening reaction.
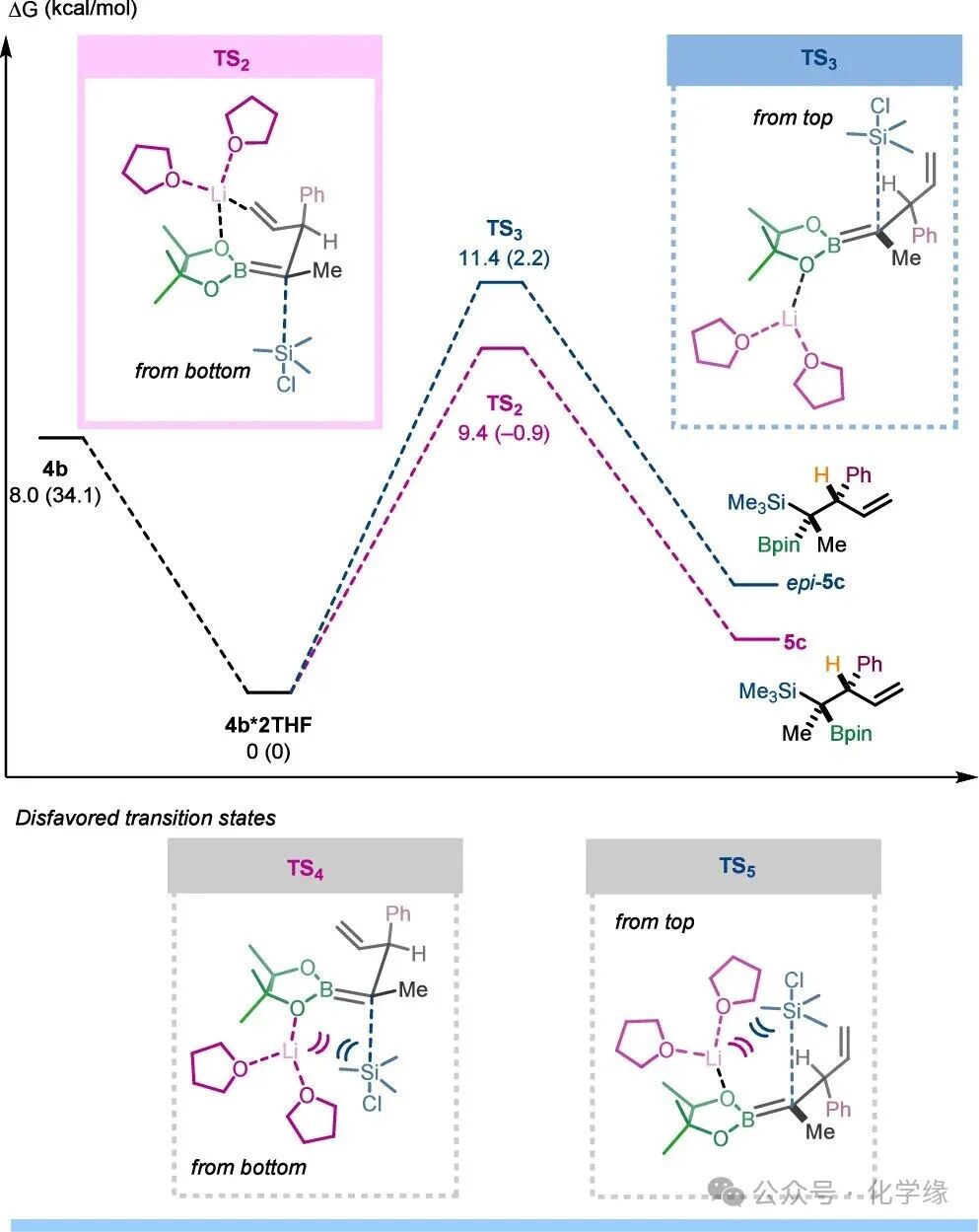
Figure 5 Silylation reaction of solvated α-boryl carbanions
Next, we turned to explore the origin of enantiomeric selectivity in the silylation step forming5c. Initially, models using free anions or implicitly solvated lithium-coordinated anions failed to match the experimental selectivity. These failures prompted us to introduce explicit solvent molecules, revealing that the formation of a double-solvated intermediate4b·2THF is highly favored (ΔΔG = −11.8 kcal/mol). Given the steric repulsion of the boronic ester is expected to have a significant impact, the model substrate incorporated a complete tertiary boronic ester.
4b·2THF‘s silylation reaction proceeds through two competitive transition statesTS2 andTS3, generating products5c (observed product) and trans-–5c. Conformational searches (using the CREST method) revealed that the electrophile approaches only from the side opposite the coordinated lithium cation (the complementary transition states areTS4 andTS5). TS2‘s calculated activation energy barrier (ΔΔG⧧ = 9.4 kcal/mol) is lower than that ofTS3 (ΔΔG⧧ = 11.4 kcal/mol) by 2.0 kcal/mol, consistent with the experimental results. This energy advantage arises from the stabilizing interaction between the lithium cation and the vinyl group inTS2, while no stabilizing interaction occurs between the lithium cation and the vinyl or phenyl groups inTS3. It is speculated that other rotamer conformations ofTS3 with lithium cation-vinyl interactions are destabilized due to steric repulsion, leading to its higher energy than the lowest energy conformation.
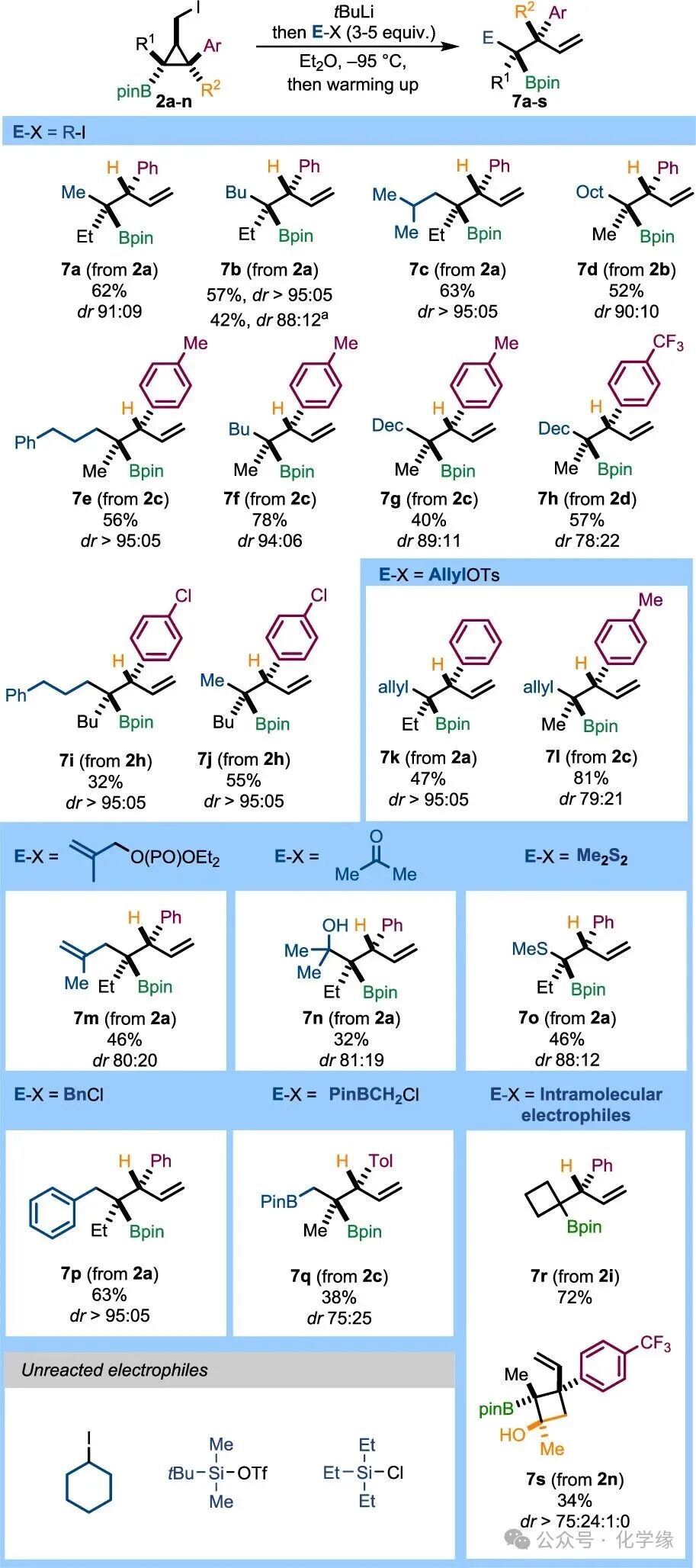
Figure 6 Substrate scope of electrophiles
To evaluate the generality of this method, a broader range of electrophiles was explored. After the ring-opening reaction of2a, the reaction with methyl iodide yielded the corresponding non-cyclic boronic ester7a with a yield of 62% and high selectivity of 91:09. Reactions with linear (R = Bu) and branched (R = sBu) primary alkyl iodides also yielded pure products, obtaining single isomers7b and7c with yields of 57% and 63%, respectively. WhenR1=Me, a slight decrease in enantiomeric selectivity was observed in reactions with primary alkyl iodides (7e–h). However, when the substituent contained electron-rich aryl substituents (phenyl or para-methylphenyl), products7d–g were obtained with high isomeric ratios and yields of 40–78%. When introducing para-fluoromethyl, the isomeric ratio further decreased, and boronic ester7h was obtained in the form of an isomeric mixture of 78:22, with a yield of 57%. In contrast, extending the alkyl chain (R1=Bu) improved isomeric selectivity, yielding single isomers of alkyl boronic esters7i and7j.
Allyl, propenyl, and benzyl reactants generated compounds7k–m and7p with yields ranging from 46% to 81% and high to excellent enantiomeric selectivity. Reaction of2a with acetone produced β-hydroxy boronic acid7n, with moderate yield and enantiomeric selectivity. Sulfur electrophiles were also applicable, generating sulfides7o with moderate yields and good isomeric selectivity. Notably, the Matteson-type homologation reaction of relatively crowded alkylation compounds was also feasible, yielding orthogonally functionalized 1,2-diboronic esters.
Additionally, several intramolecular reactions were examined. Chloride2i underwent smooth intramolecular substitution under conventional conditions without the need for external electrophiles, yielding cyclobutane7r with a yield of 72%. Finally, ketone2n exhibited complete chemical selectivity in the lithium–iodine exchange reaction.
Conclusion: Utilizing α-boryl carbanion intermediates as a platform for stereochemical generation, a series of highly stereoselective nucleophilic substitution reactions were conducted. This work pioneeringly proposes a new strategy to obtain these important intermediates from readily available stereodefined borylated cyclopropanes. The effect of the boron atom stabilizing the anion combined with stress-release-driven cleavage reactions achieved high chemical and regioselectivity, as evidenced by the broad substrate applicability and functional group tolerance. By introducing various electrophiles, ring-opened boronic esters containing vicinal tri- and tetrasubstituted chiral centers were successfully obtained. Unlike previous studies on 1,2-metal acid ester rearrangements, this transformation reflects the reactivity of formally trans-configured conversion, expanding the application scope of cyclopropyl boronic esters in stereoselective synthesis.
Article Information:
Vicinal Stereocenter Construction via α-Boryl Carbanions from Borylated Cyclopropanes
Tereza Pavlickova, Noam Orbach, Alexander Kaushansky, and Ilan Marek*
DOI: 10.1021/jacs.5c12433
We hope this article inspires you, and let us strive together for a better future!
