*For reference only by medical professionals
The latest review reports that ADC drug resistance is related to antigen expression, ADC processing, and payload efficacy.
Antibody-drug conjugates (ADC) are formed by the conjugation of specific targeting monoclonal antibodies and highly active cytotoxic drugs via linkers. With their unique pharmacological properties, ADC drugs exhibit powerful anti-tumor activity in the field of anti-tumor therapy while offering higher safety compared to traditional chemotherapy.
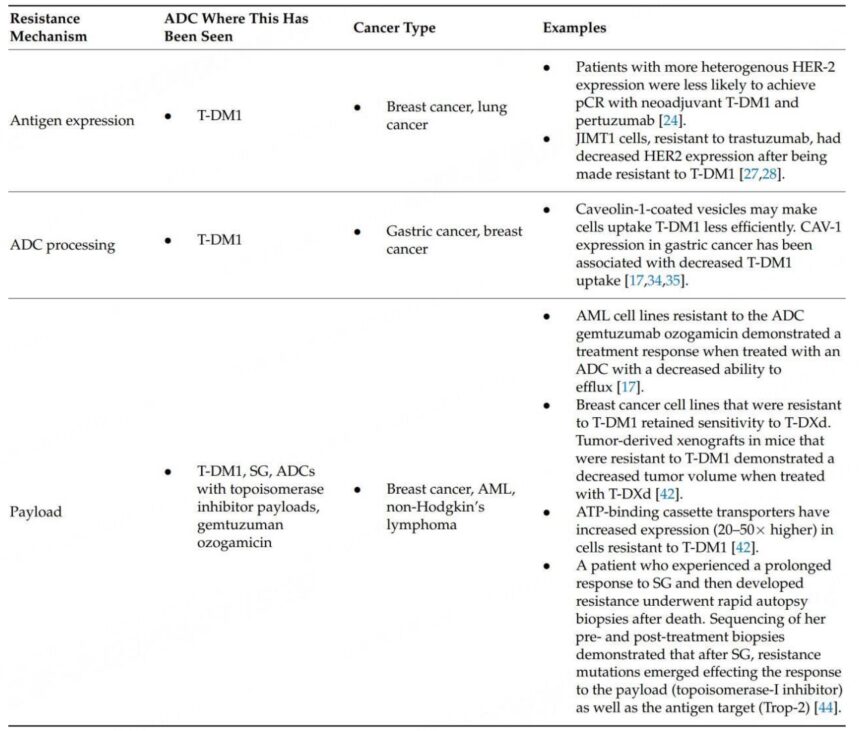
Based on the complex structure of ADCs, potential resistance mechanisms are categorized. The table below summarizes the proposed preclinical and clinical resistance mechanisms, classified according to changes in antigen expression, ADC processing, and payload efficacy. This method of categorizing resistance mechanisms provides direction for future research and helps further understand these mechanisms and new drug development targets, thereby expanding the efficacy of ADCs.
Antigen Expression
In early trials of T-DM1, tumors with higher and more uniform HER2 expression levels were more likely to respond to treatment. Considering that HER2 expression may be heterogeneous, tumor heterogeneity has been reported in 16%-36% of cases. For therapeutic drugs requiring homogeneous HER2 expression, any change in HER2 levels may lead to resistance. Further evidence supporting this hypothesis includes observations that HER2-positive tumors showed lower HER2 expression after treatment, and greater heterogeneity in expression was associated with higher recurrence rates and lower survival rates.[1] A study of early HER2-positive breast cancer patients receiving neoadjuvant treatment with T-DM1 and pertuzumab found that the presence of HER2 heterogeneity before treatment (defined as 5-50% of tumor cells with ERBB-2 amplification or HER2-FISH negative areas) could predict treatment response. In fact, among patients with heterogeneous biopsies before treatment, 0% achieved pathological complete response (pCR), whereas 55% of non-heterogeneous patients achieved pCR when treated with the combination of T-DM1 and pertuzumab.[2,3] Furthermore, circulating tumor DNA (ctDNA) from T-DM1 resistant patients showed fewer tumor cells with HER2 amplification.[4,5]
Further evidence supporting the hypothesis that decreased antigen expression mediates ADC resistance comes from preclinical cell line studies. Using JIMT1 cells, which are resistant to trastuzumab, xenograft tumors responsive to high concentrations of T-DM1 were constructed, followed by periodic T-DM1 treatment to induce resistance. Subsequent tests indicated decreased HER2 expression.[6,7] In a gastric cancer cell model, a preclinical example demonstrated the importance of the bystander effect in overcoming decreased antigen expression. These cells developed resistance to T-DM1 and then exhibited resistance to ADCs with non-cleavable linkers, while retaining sensitivity to ADCs with cleavable linkers and cell-permeable payloads.[8] Similar studies in the JIMT1 cell line identified that restoring HER2 expression could reverse resistance to T-DM1.
In addition to abnormal levels of antigen expression, dimerization of the antigen with another cell surface receptor may mediate resistance to ADCs. NRG-1β is a ligand known to induce HER2/HER3 heterodimerization, which can inhibit the cytotoxic activity of T-DM1 in HER2-amplified breast cancer cell line subpopulations. This resistance can be overcome by the addition of pertuzumab, an anti-HER2 monoclonal antibody that blocks HER2/HER3 dimerization and downstream signaling. In in vitro and in vivo tumor xenograft model studies, the combination of T-DM1 and pertuzumab showed a synergistic effect.[9] Another strategy utilizes bispecific antibodies in ADCs, involving two arms recognizing different epitopes of the target antigen. This indicates that the preclinical study of REGN5093-M114 (a bispecific MET-targeting ADC being studied in MET overexpressing EGFR mutant NSCLC cell lines) has achieved success.
T-DXd and T-DM1 have significant structural differences; T-DXd includes membrane-permeable payloads and cleavable linkers. Based on this characteristic, T-DXd has a bystander effect, where the released payload can penetrate adjacent tumor cells to exert anti-tumor effects, which is also one of the important mechanisms by which T-DXd overcomes T-DM1 resistance.
Given the impact of antigen heterogeneity in mediating ADC resistance, future strategies to address this resistance may include drugs with dual antibodies (bispecific ADCs). Additionally, combination therapies that enhance antigen expression may prove valuable. In a study of lung cancer with ERBB2 amplification or mutation, it was observed that when T-DM1 was combined with the irreversible pan-HER kinase inhibitor neratinib, lung cancer cells increased their uptake of T-DM1, whereas a decrease in T-DM1 uptake was observed with the reversible HER2 inhibitor lapatinib.
-
ADC Processing
The complexity of ADCs, especially compared to small molecules, provides many potential opportunities for resistance (Figure 1). In many cases, particularly with ADCs with non-cleavable linkers, before the ADC releases its payload, it typically binds to the target antigen, then undergoes internalization into the tumor cell, and processes intracellularly. In preclinical studies, certain reasons for resistance, particularly intracellular mechanisms, are difficult to demonstrate as driving factors for resistance, but conclusions have been drawn based on observations from therapeutic resistance models.[6] Proposed hypotheses mediating ADC resistance involve changes in intracellular uptake and processing. One proposed mechanism before ADC enters the cell involves reduced permeability to the cells through barriers, including increased cell basement membrane thickness.[6] Another proposed mechanism stems from recent preclinical work demonstrating that abnormal endosomal transport may be involved in T-DM1 resistance.[7]
One mechanism ensuring specificity for ADCs is mediated by clathrin-mediated endocytosis in cells expressing the target antigen. In the N89-TM cell line resistant to T-DM1, an alternative uptake mechanism was found, where cells utilized caveolin-1 (CAV1)-coated vesicles, which may be less efficient.[8,9] Consistent with this observation, a recent study in gastric cancer found that tumor CAV1 levels were negatively correlated with T-DM1 tumor uptake. In multiple xenograft models, genetic or pharmacological inhibition of CAV1 increased T-DM1 uptake and synergized with T-DM1.[10] Additionally, glycosaminoglycan modifications negatively regulate the internalization of tumor antigen CAIX and anti-CAIX ADC by promoting the binding of CAIX to CAV1 in membrane lipid rafts. Pharmacological inhibition of glycosaminoglycan modifications can increase internalization of anti-CAIX ADC and cytotoxic activity. In another different study, due to pH changes, a lack of proteolytic activity was observed in T-DM1 resistant cells after ADC reached the lysosome, leading to drug accumulation simulating lysosomal storage diseases.
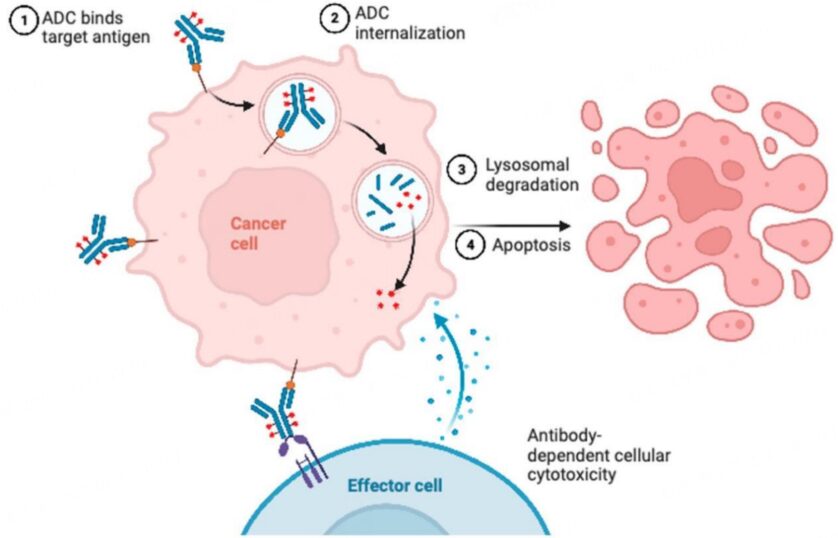
Figure 1. Potential Resistance Mechanisms of ADCs
In the JIMT1-TM cell line resistant to T-DM1, increases in proteins such as Rab5B, ATG9a, and HTT, which mediate lysosomal processing and vesicle transport, were discovered. Proteomics analysis showed particularly high protein levels of Rab6 (a protein involved in microtubule-mediated transport) and the cytoskeletal tension-related protein PAK4. To expand these findings, live-cell imaging microscopy was performed on JIMT1-TM cells and the 361-TM cell line. For the 361-TM cell line, compared to parental cells that are not resistant to T-DM1, ADCs with non-cleavable linkers spent significantly longer time in the lysosome, raising awareness about the lower efficiency of ADC processing. Interestingly, this result was not observed with ADCs containing cleavable linkers, indicating mechanisms to overcome resistance. In the JIMT1-TM cell line, both ADCs with cleavable and non-cleavable linkers showed longer co-localization in the lysosome than parental cells. Unsurprisingly, compared to parental cells, both resistant cell lines showed reduced levels of linker-payload metabolites, but it remains uncertain whether this is driven by decreased HER2 expression, reduced ADC processing, or both. Consistent with the view that payload release may involve different ADCs depending on linker status, a recent study using CRISPR screening demonstrated that ADCs with cleavable linkers are rapidly processed immediately after ADC uptake and transported to early endosomes, while ADCs with non-cleavable linkers require further lysosomal delivery to successfully release their payload. Additionally, literature reports that depletion of sialic acid sensitizes cells to ADC toxicity by enhancing lysosomal delivery of ADCs (possibly by reducing ADC recycling).[11]
-
Payload
Some observed resistance mechanisms involve the payload itself, which can be overcome by using ADCs with alternative payloads. For example, ADC resistance to topoisomerase inhibitors may be driven by changes in topoisomerase expression or downstream signaling mechanisms (Figure 2). This has also been observed in tumor models of non-Hodgkin lymphoma, where replacing ADCs containing auristatin payloads with those based on anthracycline derivatives helped patients further respond to ADC therapy. Similarly, in breast cancer cells resistant to T-DM1, tumor cells remained sensitive to T-DXd. To track these results, xenograft mouse models were established with T-DM1 sensitive and resistant cells. The xenograft models constructed from T-DM1 resistant cells were insensitive to T-DM1, but when treated with T-DXd, these mice showed reduced tumor volume.[12] The application of this theory includes the development of other HER2-targeted or TROP-2-targeted ADCs, which may provide alternative payloads to overcome payload-related resistance.

Figure 2. Resistance Mechanisms of ADCs
The ability to perform whole exome sequencing during patient ADC treatment allows real-time tracking of the development of resistance mutations and provides theoretical support for future treatments. For example, a study assessed the resistance mechanisms of a patient who had a prolonged response to SG (over 8 months) and underwent whole exome sequencing before starting treatment. After progression, rapid autopsy sampling was conducted, and treatment samples were compared before and after with SG. Analysis of mutations that occurred after treatment indicated that tumor subclones carried TOP1 mutations (encoding topoisomerase-1, the target of SG payload) and unique subclones carried TACSTD2 mutations (encoding TROP-2, the antigen target of SG). These findings suggest parallel resistance mechanisms targeting ADC antibodies and payloads, indicating that tumor cells may become resistant to sequential ADCs, including those with similar payloads or similar antibody targets.[13] Future innovations in targeted therapies and rapid sequencing may provide personalized treatment guidance based on resistance mutations generated from previous treatments.
Increased efflux of ADC payloads mediated by ATP-binding cassette transporters is another potential resistance mechanism. Compared to parental cells, increased expression of transport proteins was observed in T-DM1 resistant cells, up to 20-50 times. This mechanism was observed in AML cell models as a means of resistance to gemtuzumab ozogamicin (GO). Similarly, patients with lower levels of ABCB1, which encodes ATP-binding cassette, showed improved responses to GO. In different cell lines, after developing resistance to T-DM1, expression of the gene ABCC1 increased. Lastly, in preclinical models of paclitaxel-resistant cancer cell lines, ADC resistance to paclitaxel-derived payloads was associated with ABCB1 overexpression-mediated TUBB3 activation.
Multiple potential preclinical and clinical mechanisms have been proposed to explain these resistance strategies. Studies have found that T-DM1 resistant cell lines also exhibit resistance to trastuzumab-guided ADCs with non-cleavable linkers. When the drug efflux pump ABCC1 is reversed or knocked down using siRNA, tumor cells show new sensitivity to ADCs containing non-cleavable linkers. Interestingly, in GO-resistant AML cells, new remissions occurred when using the less efflux-potential cytotoxic drug vadastuximab talirine instead. After 9 months of ADC treatment, RNAseq analysis was performed in preclinical breast cancer mouse models treated with an anti-nectin-4 targeted ADC (named N41mab-vc-MMAE). Furthermore, in this model, upregulation of ABCB1 was observed. When ADCs were combined with the pharmacological inhibitor tariquidar, rapid therapeutic responses were observed, with tolerability significantly better than that of tariquidar and docetaxel combination treatment.[14]
