

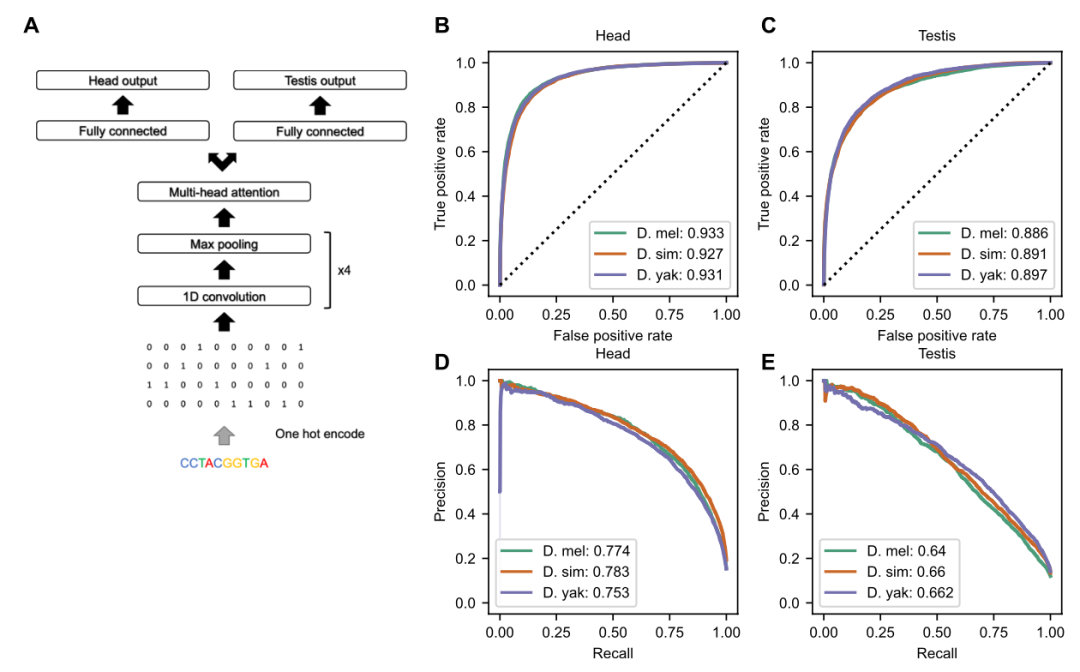
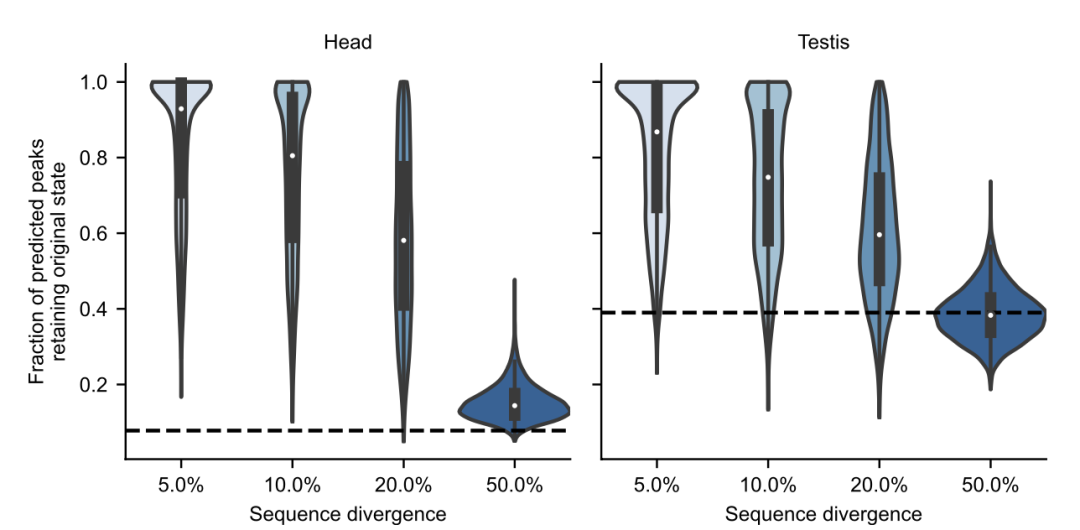
Editor: Eleven





Reprint Notice
This article is not original. The copyright belongs to the author. Personal forwarding and sharing are welcome; reprinting without the author’s permission is prohibited. The author retains all legal rights, and violators will be prosecuted.
