
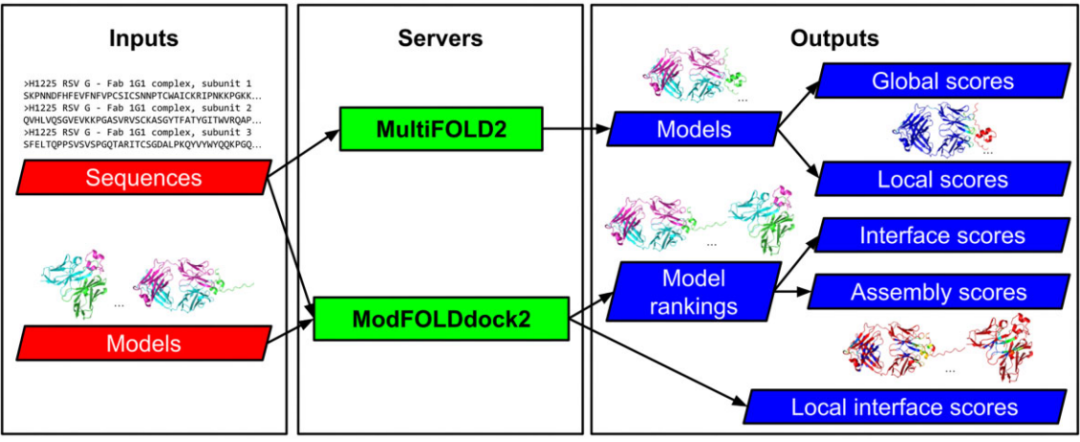
/ Stronger than AlphaFold3? MultiFOLD2 and ModFOLDdock2: The new kings of protein complex structure prediction and assessment! /
With AlphaFold2 revolutionizing the field of protein structure prediction, the scientific community has quickly turned its attention to more complex challenges —the precise modeling of protein complexes (quaternary structures). While monomer predictions are nearly perfect, the assembly of oligomers, especially how to predict the correctstoichiometry, interface interactions, and overall assembly quality remain full of unknowns.
In this competitive research landscape, a research team from the University of Reading’s School of Biological Sciences has launched two powerful servers —MultiFOLD2: focused on protein complex structure predictionModFOLDdock2: focused on assessing the quality of complex models.
 Click to view the original textMultiFOLD2 ModFOLDdock2.pdf
Click to view the original textMultiFOLD2 ModFOLDdock2.pdf
They not only performed excellently in the latest International Structure Prediction CompetitionCASP16, but also surpassed several strong competitors, includingAlphaFold3, in the real-time independent evaluation of CAMEO. Today, we will take a detailed look at how these two next-generation tools have become the new kings of protein quaternary structure prediction and validation!
Why do we need new tools for quaternary structure?
Although AlphaFold2 has significantly improved the accuracy of monomer predictions, existing methods still have obvious shortcomings when it comes to multi-chain complexes:
Number and ratio of components (Stoichiometry) are often unknown.
Inter-chain interface interactions are difficult to model accurately.
Quality assessment of predicted models lacks reliable methods..
To address these issues, the University of Reading team has made significant upgrades based on the previous MultiFOLD and ModFOLDdock, launchingMultiFOLD2 and ModFOLDdock2.
MultiFOLD2: A Smarter Complex Modeler
One of the highlights of MultiFOLD2 is: even if users do not know the stoichiometry of the complex, it canautomatically infer stoichiometry and perform structure predictions based on this information.
The complete process includes three major steps:
Sampling
Integrates various strategies including LocalColabFold (based on AlphaFold2), AlphaFold2-Multimer (with dropout), RoseTTAFold2, and RoseTTAFold-All-Atom.
Scoring
Uses ModFOLDdock2S for unified scoring and model selection.
Refinement
Further applies the AlphaFold2-Multimer_Refine process to the top 5 models to enhance accuracy.
Even more impressively, MultiFOLD2 outperformed AlphaFold3 in overall performance during the CAMEO real-time evaluation, becoming one of the strongest complex modeling servers currently available.
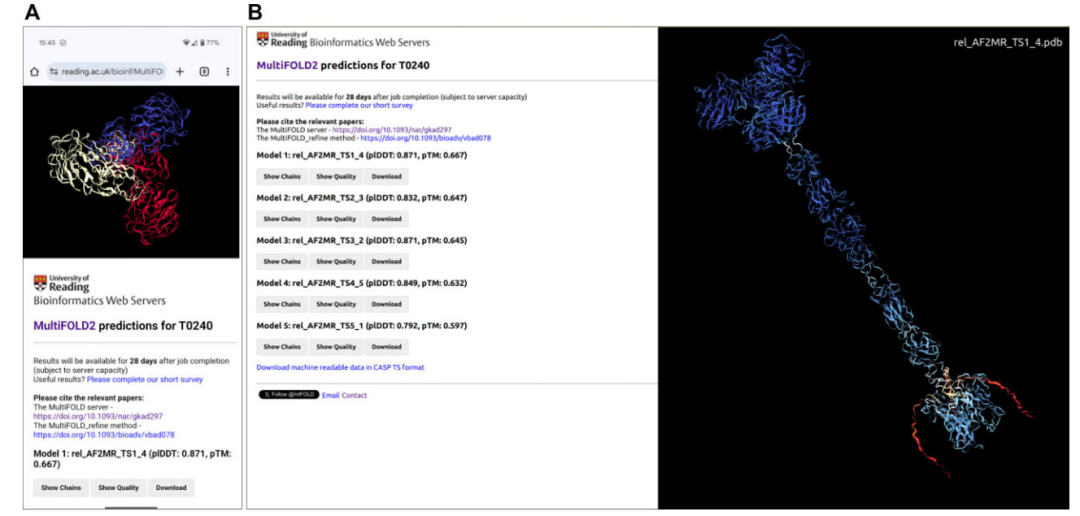 MultiFOLD2 server results pages for CASP16 target T0240 with correctly predicted stoichiometry A3, viewed using mobile and desktop browsers
MultiFOLD2 server results pages for CASP16 target T0240 with correctly predicted stoichiometry A3, viewed using mobile and desktop browsers
ModFOLDdock2: Making Model Quality Clear at a Glance
ModFOLDdock2 is responsible for systematic quality assessment of predicted complex structures. It can provide:
Global assembly quality score (overall structural accuracy)
Local interface residue scores (accuracy of inter-chain contact points)
ModFOLDdock2 offers three usage modes:
ModFOLDdock2: Multi-model evaluation, optimizing the linear correlation between scores and actual quality.
ModFOLDdock2R: Multi-model evaluation, optimizing model ranking.
ModFOLDdock2S: Single model evaluation, suitable for cases with only one predicted model.
More importantly, local interface quality assessment introduces neural networks (NN), significantly improving the accuracy of fine-grained interface predictions.
In the international blind test competitionCASP16, ModFOLDdock2 won first place in the interface quality scoring project (QSCORE and local accuracy)!
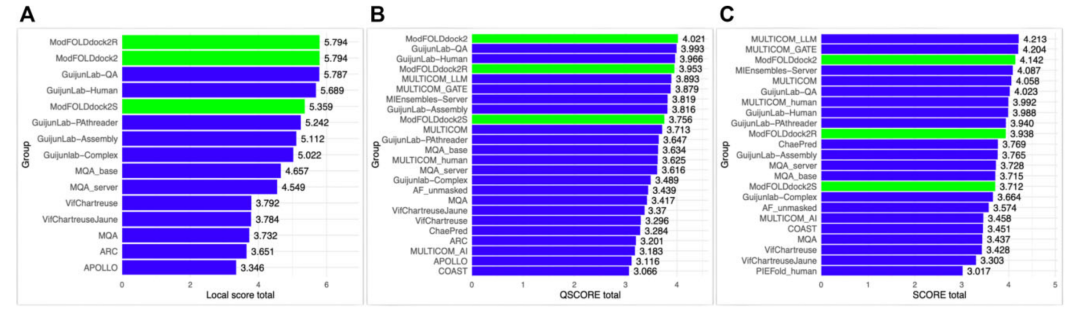 The performance of the ModFOLDdock2 server methods in CASP16 versus other participating predictor groups according to the official scores
The performance of the ModFOLDdock2 server methods in CASP16 versus other participating predictor groups according to the official scores
Two Swords Combined, Outstanding Achievements
Overview of Independent Evaluation Results:
CAMEO Continuous Testing: MultiFOLD2 significantly outperformed AlphaFold3 in lDDT scores across various targets (monomers, oligomers).
CASP16 International Blind Test:
MultiFOLD2 ranked first on the hardest targets, overall in the top six, comprehensively surpassing AF3.
ModFOLDdock2 won first place in interface quality scoring, with overall structural scoring also consistently ranking high.
Not only is the accuracy high, but the server interface is also very user-friendly, supporting browser-based 3D interactive viewing and providing machine-readable data in standard formats, making it convenient for researchers to directly interface with downstream analyses.
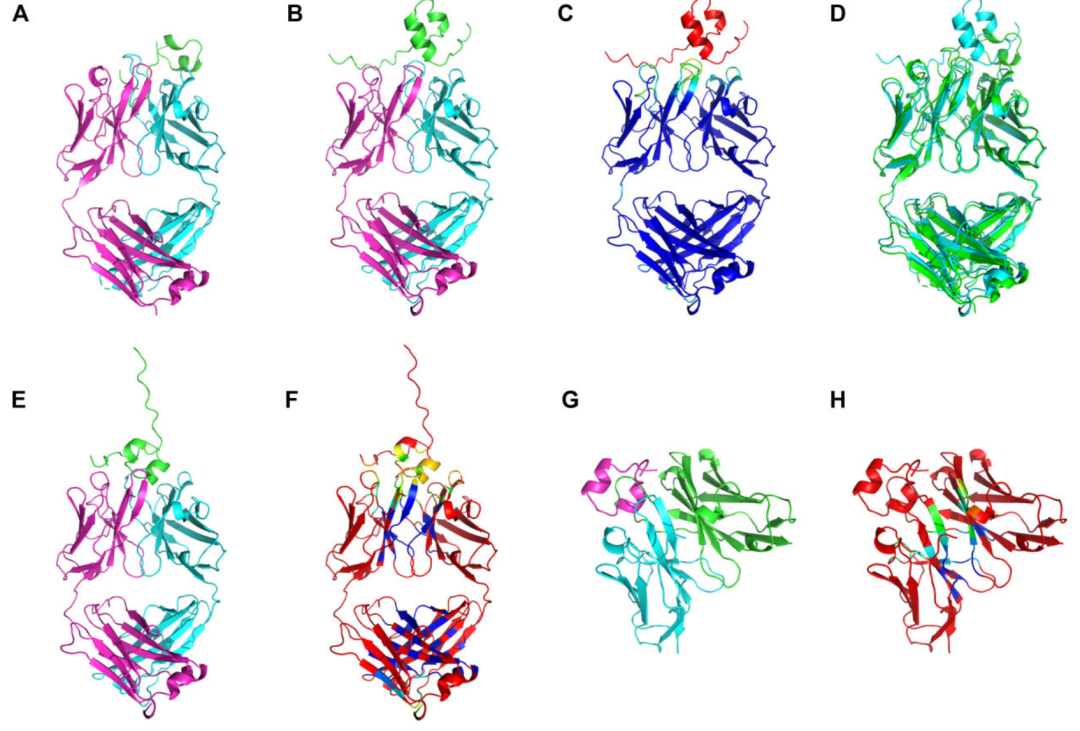 Examples of MultiFOLD2 and ModFOLDdock2 predictions for a heteromultimeric CASP16 target H0225 (Phase 0)/H1225 (Phase 1) with the stoichiometry A1B1C1
Examples of MultiFOLD2 and ModFOLDdock2 predictions for a heteromultimeric CASP16 target H0225 (Phase 0)/H1225 (Phase 1) with the stoichiometry A1B1C1
How to Use?
MultiFOLD2 and ModFOLDdock2 are both now freely available to the global community:
🧩MultiFOLD2 Prediction Server: Access MultiFOLD2 ( https://www.reading.ac.uk/bioinf/MultiFOLD/ )
🔍ModFOLDdock2 Evaluation Server: Access ModFOLDdock2 ( https://www.reading.ac.uk/bioinf/ModFOLDdock/ )
🐳Docker Image: Download from Docker Hub ( https://hub.docker.com/r/mcguffin/multifold2 )
Whether upgrading from monomer predictions to complexes or wanting to independently validate the quality of constructed oligomer models, you can take off with just one click!
In the era of AI accelerating structural biology, accurate, efficient, and user-friendly protein complex prediction tools are crucial. MultiFOLD2 and ModFOLDdock2 are providing powerful support for researchers with their outstanding performance, opening up new horizons for protein interaction and function research.
In the future, as more structures of biological macromolecular complexes are deciphered, we have reason to believe that this golden duo will continue to lead a new wave in quaternary structure modeling!
🧠 Feel free to scan the QR code to add the editor and join the group for discussions, tracking the cutting-edge developments in structural biology intelligence!