Hello everyone, today I would like to share an article published in the Journal of the American Chemical Society, titled Dual Active Sites on Cu/Cu2O Heterostructures for the Cascade Electrocatalytic Synthesis of Amino Acids. The corresponding author of the article is Professor Jianlin Shi from the Shanghai Institute of Ceramics, Chinese Academy of Sciences.

Abstract
Electrocatalytic C-N coupling enables the efficient and sustainable production of amino acids. However, it is affected by complex reaction pathways and intense adsorption competition between reactants and intermediates at a single active site. This paper reports a cascade catalytic strategy on Cu/Cu2O heterostructures for the electrosynthesis of amino acids from nitrates and keto acids. These catalysts can produce various amino acids (including alanine, glycine, leucine, and glutamic acid) with a Faradaic efficiency of up to 75.78% and an alanine yield of up to 478.89 mmol h-1g cat-1. Mechanistic exploration reveals the reconstruction of CuO/C electrocatalysts into Cu/Cu2O heterostructures and a cascade catalytic pathway on Cu0/Cu+ dual sites. The reduction of NO3– and the protonation of the C=N intermediate are sequentially achieved at the Cu0 and Cu+ sites, respectively. Meanwhile, the rich Cu/Cu2O interface facilitates the transfer of intermediates. This work provides new insights into the design of bimetallic active sites for the electrosynthesis of amino acids, promoting the efficient and sustainable production of high-value organic nitrogen compounds.
Introduction
Biological synthesis and chemical synthesis are the two main pathways for producing amino acids. Biological fermentation is the primary industrial method, but it often uses high-cost raw materials and complex purification processes. Compared to biological fermentation, chemical synthesis significantly reduces production costs but requires the use of highly toxic cyanides and energy-intensive ammonia (NH3). In contrast, electrocatalytic carbon-nitrogen coupling between abundant carbon feedstocks and nitrogen waste provides a more efficient and environmentally friendly synthesis strategy for amino acid synthesis, capable of simultaneously removing environmental pollutants and producing organic nitrogen compounds.
Nam et al. first achieved the electrosynthesis of amino acids from oxalic acid and nitrates (NO3−) on a copper-mercury electrode, with a Faradaic efficiency (FE) of 43.1%. However, the reaction pathways for electrocatalytic coupling to form C-N bonds under the aforementioned environmental conditions are complex and difficult to control, involving multiple electron and proton transfers, as well as many competing side reactions, such as the hydrogenation of carbonaceous reactants, deep reduction of NOx to NH3, and hydrogen evolution reactions. In recent years, researchers have explored various strategies to address these challenges from different perspectives. Using a self-supporting membrane with CoFe alloy (CoFe-SSM) catalysts to leverage the metal synergy effect and effective mass transfer on the self-supporting membrane, successful synthesis of leucine was achieved through the co-reduction of α-keto acids and NO, with a FE of 32.4% and a yield of 115.4 μmol h-1. The Zhang team used oxide-derived Ag catalysts with low-coordination sites in two decoupled flow reactors to reduce by-products during the slow reduction of acetone oxime, thus achieving a FE of 70% for α-keto acids and NO, producing alanine in gram quantities. On PdCu nanowires, a possible tandem electrochemical-chemical-electrochemical cascade process achieved a 54.8% FE for alanine. To effectively suppress hydrogen evolution reactions, hydrophobic groups were introduced on a crystalline organic framework (F-Cu3-OF) catalyst containing tri-nuclear copper clusters, constructing short-distance copper atoms that simultaneously activate NO3– and keto acids, thus synthesizing a series of amino acids under alkaline conditions. Cobalt phthalocyanine (CoPc/CNT) catalysts immobilized on carbon nanotubes were applied for one-pot synthesis of various amino acids, achieving an FE of 61% for alanine. Based on the above studies, the authors found that on common single active sites, the intense adsorption competition between reactants and C=N intermediates leads to low activity (yield) and selectivity (FE) for amino acids. In this regard, this paper aims to design dual active sites to balance the adsorption of reactants and intermediates, thereby optimizing the reaction pathway for C-N coupling.
In this work, the authors fabricated CuO/C electrocatalysts and obtained a Cu/Cu2O heterostructure with dual metallic active sites through an electrochemical reconstruction process. On this heterostructure, under ambient conditions, various amino acids including alanine, glycine, leucine, and glutamic acid were synthesized through the C-N coupling between keto acids and NO3– in acidic electrolytes. In alanine production, at -0.6 V (vs RHE), the catalyst provided a significant enhancement of 75.78% in FE, and at -0.7 V (vs RHE), the yield reached up to 478.89 mmol h-1g cat-1, with excellent stability over at least 30 h of continuous reaction. Experimental and density functional theory (DFT) calculations reveal the cascade catalytic mechanism for amino acid synthesis on Cu/Cu2O heterostructures. The Cu/Cu2O heterostructure possesses Cu0/Cu+ dual active sites. At the Cu0 site, keto acids and NO3– preferentially adsorb and couple to form the C=N intermediate, which then migrates to the nearby Cu+ site for protonation, producing *CH3CNHCOO–. The rich Cu/Cu2O interface facilitates the transfer of the C=N intermediate, enhancing the reaction rate and assisting the cascade catalysis on the Cu0/Cu+ dual active sites, achieving excellent performance.
Results and Discussion
Structural Characterization of Cu/Cu2O Heterostructures
The CuO/C electrocatalyst was synthesized through a mild heating reaction, followed by a simple pyrolysis process (Figure 1a). The Cu metal-organic framework HKUST1 was first assembled by mixing Cu2+ and trimesic acid (H3BTC) in methanol at 60℃. As depicted in high-resolution transmission electron microscopy (HRTEM) images, the CuO/C800 catalyst underwent reconstruction into the Cu/Cu2O heterostructure after electrocatalysis, with a rich interface between Cu (111) and Cu2O (111) facets (Figure 1a, b). Energy-dispersive X-ray spectroscopy (EDS) elemental mapping revealed a uniform distribution of Cu, C, and O elements in these catalysts (Figure 1c). X-ray diffraction (XRD) patterns confirmed the reduction of CuO to Cu/Cu2O during the electrocatalytic process (Figure 1d). Under the same reaction conditions, the content of Cu2O exhibited an opposite dependence on the pyrolysis temperature, which may be due to the suppression of Cu2O further reduction to Cu. In the CuO/C800 Cu 2p X-ray photoelectron spectroscopy (XPS) analysis (Figure 1e), characteristic peaks at 933.7 and 953.8 eV were assigned to Cu2+ species. After the electrochemical reaction, two new peaks appeared at 932.8 and 952.7 eV, corresponding to the electro-reduction of Cu0/Cu+ mixed state. The Auger electron spectroscopy (AES) analysis of the CuO/C catalyst revealed characteristic peaks of Cu0 and Cu+, showing that the content of Cu2O decreased with increasing pyrolysis temperature, which is consistent with the XRD results. More intuitively, the in situ Raman spectra of the electrode during the electrochemical reaction at negative potentials (Figure 1g) showed that the CuO/C800 phase almost completely disappeared, and then after 10 minutes, the Cu2O phase gradually appeared, revealing the reconstruction from CuO to Cu/Cu2O heterostructures, consistent with the HRTEM, XRD, and XPS results.
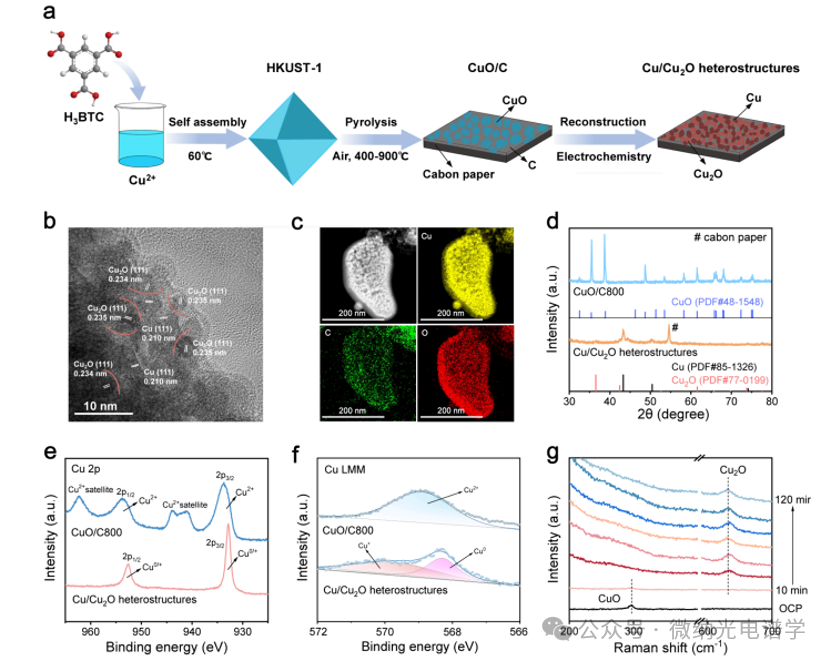
Figure 1. Synthesis and characterization of the CuO/C800 catalyst and its derived Cu/Cu2O heterostructure. (a) Schematic diagram of the synthesis of the CuO/C800 catalyst and Cu/Cu2O heterostructure. (b) HR-TEM images of the Cu/Cu2O heterostructure and (c) EDS mapping images. (d) XRD patterns of the CuO/C800 catalyst and Cu/Cu2O heterostructure, (e) XPS Cu 2p spectra and (f) AES Cu LMM spectra. (g) In situ Raman spectra of the CuO/C800 catalyst during the electrocatalytic synthesis of alanine.
Electrocatalytic Synthesis of Amino Acids
The electrocatalytic synthesis of amino acids was conducted under ambient conditions in an H-type electrolyzer, with pyruvic acid (PA) and potassium nitrate (KNO3) serving as carbon and nitrogen sources, respectively. The liquid-phase products were analyzed using liquid chromatography-mass spectrometry (LC-MS) (Figure 2a). The electrocatalytic C-N coupling activity of CuO/C was evaluated through linear sweep voltammetry (LSV) measurements in Ar-saturated electrolytes (Figure 2b). Compared to hydrogen evolution reactions (HERS), pyruvic acid reduction reactions (PARR), and the reduction reactions of NO3 (NO3RR), the current density of the C-N coupling reaction of NO3– and PA significantly increased. Chronoamperometry was used to study the potential dependence and further evaluate the electrocatalytic activity of CuO/C in alanine synthesis over 2 h (Figure 2c). From -0.3 to -0.7 V (vs RHE), the FE of alanine on the CuO/C800 catalyst varied with the applied potential, reaching a maximum of 75.78% at -0.6 V (vs RHE). When the applied potential increased to -0.7 V (vs RHE), the yield of alanine was 478.89 mmol h-1g cat-1. However, a small amount of oxime was detected in the electrolyte, indicating that NH2OH may also participate in the C-N coupling reaction. The effect of the NO3–/PA ratio and the concentration of NO3– on alanine production was studied. Figure 2d reveals that the highest FE for alanine was achieved at a molar ratio of 2.5:1 for NO3–/PA. At this ratio, the total FE of the products approached 1, with negligible generation of NH3 and H2, indicating the formation of the preferred target product.
Control experiments were conducted to determine the key active sites for amino acid electrosynthesis in the CuO/C catalyst. Compared to CuO/C400, CuO/C600, and CuO/C900, the CuO/C800 catalyst exhibited enhanced performance in alanine electrosynthesis, attributed to its optimal carbon content at this pyrolysis temperature, which facilitated the formation of a Cu/Cu2O heterostructure with a rich interface, thereby improving the production efficiency of alanine (Figure 2g). Figure 2g also shows that the current density decreased with decreasing carbon content, indicating that carbon species contribute to charge transfer during the electrocatalytic process. Based on the above, the unique Cu/Cu2O heterostructure derived from the CuO/C800 catalyst plays a key role in the electrosynthesis of amino acids.
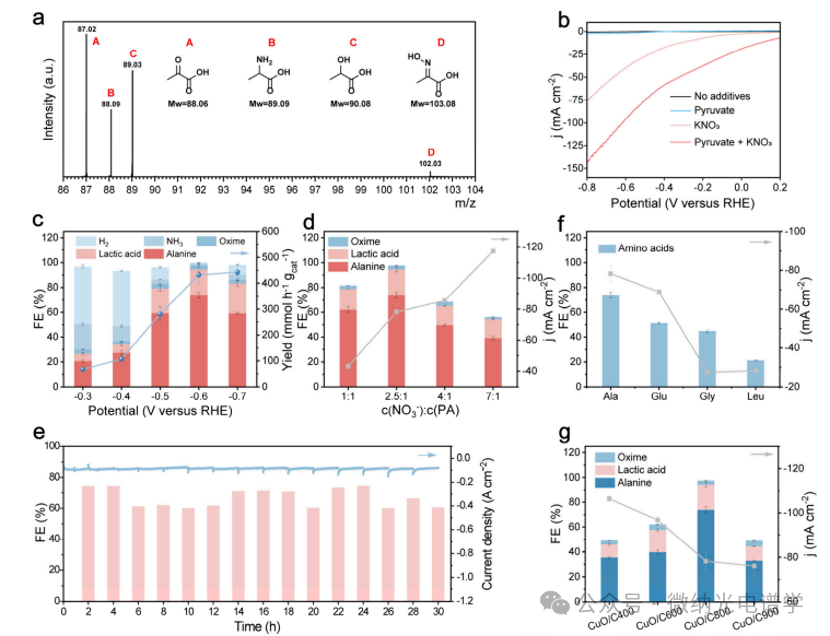
Figure 2. Detection and electrosynthesis of amino acids. (a) LC-MS spectra of products for qualitative identification. (b) LSV curves of the CuO/C800 catalyst in different electrolytes, with a scan rate of 0.05 V/s. (c) Potential-dependent FEs and yields of alanine on the CuO/C800 catalyst in 0.5 M KNO3 at 0.2 MPA. (d) FEs and current densities of alanine in different electrolytes with varying NO3– PA molar ratios at -0.6 V and RHE. (e) Long-term stability test of the CuO/C800 catalyst for alanine electrosynthesis in 0.5 M KNO3 at -0.6 V and RHE. (f) Optimized FEs of alanine, glutamic acid, glycine, and leucine and their corresponding current densities on the CuO/C800 catalyst. (g) Maximum FEs of alanine and their corresponding current densities on the CuO/C400, CuO/C600, CuO/C800, and CuO/C900 catalysts.
In Situ Characterization to Explore Reaction Pathways
From Figure 3a, the time-resolved Raman spectra of the CuO/C800 catalyst recorded at -0.6 V vs RHE show that the vibrational peak at 1176 cm-1 is attributed to the swinging mode of the *NH2 species corresponding to the formation of key N- intermediates. After 30 min of electrochemical reaction, the stretching vibrations of C=N (1717 cm-1) and C-N (1393 cm-1) appeared, verifying the coupling of PA and *NH2 to form the C=N containing intermediates and alanine. The time-resolved SR-FTIR spectra of the CuO/C800 catalyst, as shown in Figure 3b, show that the swinging mode of *NH2 at 1247 cm-1 marks the formation of *NHx species from NO3RR. Meanwhile, the characteristic band at 1593 cm-1 was assigned to the stretching mode of C=N, verifying the coupling reaction of *NH2 and PA. Additionally, a C-N bond located at 1412 cm-1 was observed, proving the formation of alanine. During the electro-reduction process, signals of *NH2, C=N, and C-N bonds were also observed on the Cu catalyst, but these signals were significantly weaker than those on the CuO/C800, consistent with the electrochemical experimental results. Due to the high sensitivity of infrared spectroscopy (IR) to the vibrational modes of polar groups, the adsorption sites of *NH2 and C=N bonds can be identified by analyzing the positions of the characteristic IR bands. Notably, the IR bands corresponding to *NH2 on the CuO/C800 and Cu catalysts are located at the same position, while the IR bands associated with the C=N bond on the CuO/C800 are shifted compared to the corresponding bands on the Cu catalyst. This indicates that the adsorption of *NH2 occurs at the Cu0 site, while the intermediates containing C N bonds are primarily located at the Cu+ site. Based on the above results, the authors propose a possible dominant cascade reaction pathway for the coupling of NO3– and PA to form alanine (Figure 3e). The coupling of PA with the NO3RR generated *NH2 at the Cu0 site, producing C=N species, which then migrate to the Cu+ site and are ultimately reduced to the desired alanine.
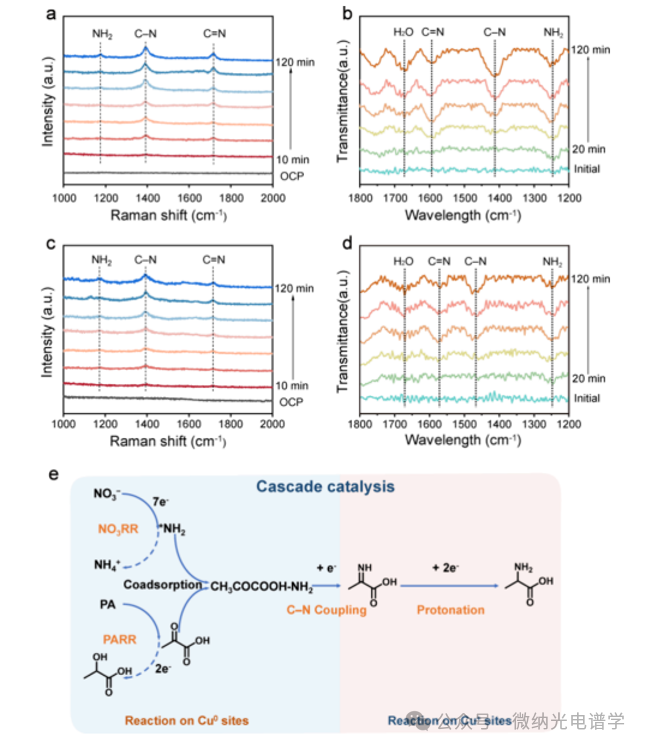
Figure 3. Reaction pathways for alanine generation. (a) Time-resolved in situ Raman spectra of the CuO/C800 catalyst during alanine synthesis and (b) in situ SR-FTIR spectra. Time-resolved in situ Raman spectra of the Cu catalyst during alanine synthesis and (d) in situ SR-FTIR spectra. (e) Schematic diagram of the coupling of PA and NO3– to form alanine on the Cu/Cu2O heterostructure.
Theoretical Calculations of Cu/Cu2O Heterostructures
To further investigate the C-N coupling reaction pathways on Cu/Cu2O heterostructures, density functional theory (DFT) calculations were performed by constructing a Cu (111)/Cu2O (111) heterostructure model. The Gibbs free energies (ΔG) of the adsorption of NO3–, CH3COCOO–, and CH3CNHCOO– on the Cu/Cu2O heterostructure were calculated (Figure 4a-c). The adsorption of NO3– and CH3COCOO– is more favorable on Cu (111) than on Cu2O (111), while CH3CNHCOO– prefers to adsorb on Cu2O (111). Therefore, the active sites for the reduction of NO3– and C=N intermediates were calculated for the entire reaction pathway using Cu (111) and Cu2O (111) as the respective active sites. As shown in Figures 4d, e, the two steps *NO→*NHO and *NHO→*NHOH are thermodynamically unfavorable, with uphill ΔG of +0.2 and +0.14 eV, identified as the rate-determining steps (RDSs). Compared to the formation of *NH2OH, the generation of *NH exhibits a lower Gibbs free energy barrier (ΔG=-2.48 eV), making *NH2 thermodynamically superior to *NH2OH. Comprehensive electrochemical, spectroscopic, and DFT studies confirm that the NH2 mediated pathway is the main reaction pathway, while the NH2OH mediated pathway is a possible accompanying process. Notably, due to the hydrogen bonding interactions between *CH3COCOO– and *NH2, the formation of *CH3COCOO–-NH2 is more favorable, with a downhill ΔG of -1.77 eV. In contrast, the conversion of *NH2 to *NH3 is thermodynamically unfavorable, with an uphill ΔG of +0.04 eV. Subsequently, *CH3COCOO– and *NH2 spontaneously undergo C-N coupling, forming *CH3CNHCOO– with a free energy of -9.24 eV. Subsequently, *CH3CNHCOO– migrates to the adjacent Cu+ site and is further reduced to form *CH3CHNH2COO–. The DFT results indicate that Cu and Cu2O exhibit cascade catalytic effects during the reaction process, with the Cu0 site facilitating the activation of NO3– and C-N coupling, while the Cu+ site accelerates subsequent hydrogenation steps.
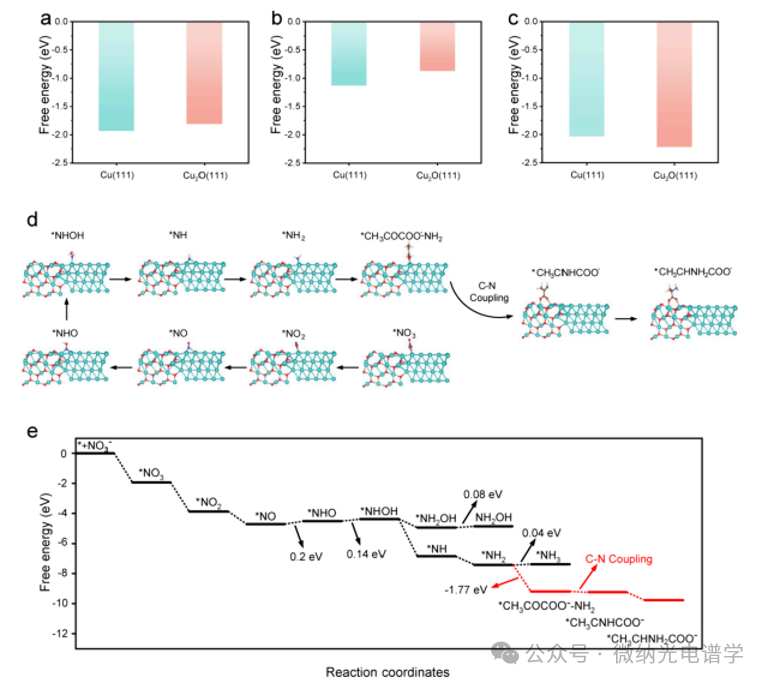
Figure 4. DFT calculations of the Cu (111)/Cu2O (111) heterostructure model. (a) Adsorption free energies of NO3–, (b) CH3COCOO–, and (c) CH3CNHCOO– on the Cu (111) and Cu2O (111) facets. (d) Atomic configurations and (e) corresponding free energies for alanine synthesis on the Cu (111)/Cu2O (111) heterostructure.
Conclusion
A cascade catalytic route is demonstrated on the Cu/Cu2O heterostructure for the effective synthesis of amino acids from NO3– and keto acids. The composite catalyst can catalyze the synthesis of alanine, glycine, leucine, and glutamic acid, with an ultra-high FE of 75.78% and a yield of up to 478.89 mmol h-1gcat-1. It is revealed that the reduction of NO3– and the formation of C=N bonds are attributed to the Cu0 site, while the protonation of the C=N intermediate often occurs at the nearby Cu+ site. Cascade catalysis facilitates the adsorption of reactants (NO3– and CH3COCOO–) and intermediates (*CH3CNHCOO–) on the Cu and Cu2O sites, respectively, promoting the synergistic C-N coupling, thereby enhancing the yield and selectivity of amino acids. This work not only provides a cascade catalytic route to improve the yield and selectivity of amino acids but also offers an alternative strategy for designing dual-site electrocatalysts to effectively construct C-N bonds.
Original link: https://doi.org/10.1021/jacs.5c04649
Editor: Liang Cailin
Reviewer: He Yiyang