Atypical hemolytic uremic syndrome (aHUS) is a type of complement-mediated thrombotic microangiopathy (TMA), characterized by microangiopathic hemolytic anemia (MAHA), thrombocytopenia, and multi-organ damage as typical manifestations, with infection and pregnancy being the main triggers.[1]
The condition of aHUS is usually quite critical. In the pre-complement treatment era, the mortality rate during the acute phase was 25%, and about 50% of patients progressed to end-stage renal disease within one year.[1] Rapid and accurate diagnosis and timely treatment are crucial for the prognosis of aHUS patients.
However, most aHUS patients have triggering factors or concomitant diseases, making diagnosis and differential diagnosis more challenging.[2]
This article will provide a detailed analysis of the pathogenesis, diagnosis, differential diagnosis, and treatment strategies of this rare “uremia” for clinical reference.
01
Pathogenesis: Interaction Between Genetic Factors and Triggers Leading to Onset
1.1 Uncontrolled Complement Regulatory Factors Trigger TMA, Leading to Multi-Organ Damage
aHUS is a rare TMA caused by abnormalities in complement bypass pathway regulatory proteins, including hereditary and acquired complement regulatory dysfunction. The former is mainly due to mutations in complement-related pathogenic genes such as complement factor H (CFH), complement factor I (CFI), membrane cofactor protein (MCP), and complement C3 (C3), while the latter is related to the production of anti-factor H antibodies.[3]
In the bypass pathway, complement C3 can lead to the formation of the membrane attack complex (C5b-9) through a series of complement and regulatory factor reactions, with C5b-9 further forming membrane pores that can directly lyse pathogens or target cells. In aHUS patients, mutations in complement regulatory factors trigger uncontrolled complement activation. Persistent complement overactivation leads to endothelial cell damage, hemolysis of red blood cells, platelet activation, increased coagulation and thrombosis, the occurrence of TMA, and ultimately causes multi-organ damage in the kidneys, brain, heart, lungs, gastrointestinal tract, etc.[4-6]
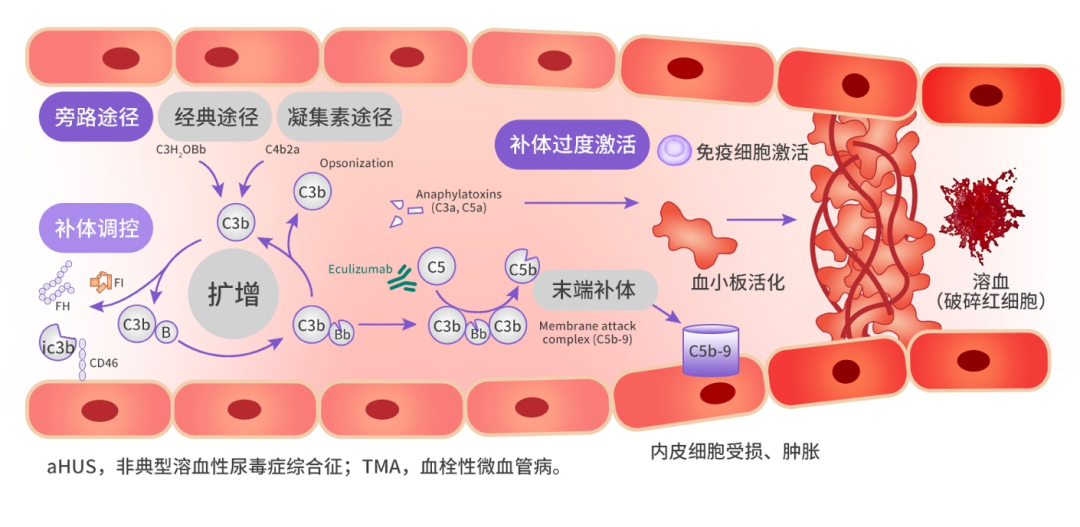
Figure 1: Schematic Diagram of aHUS Pathological Mechanism[7,8]
1.2 Multiple Triggers and Overactivation of Complement Interact to Promote the Development of aHUS
The onset of aHUS depends on the interaction between the patient’s genetic predisposition and environmental triggering factors. Studies show that 69% of aHUS patients with genetic complement abnormalities had triggering diseases/conditions present early on.[2]
Triggering factors include autoimmune diseases, transplantation, hypertension, pregnancy, medications, infections, tumors, etc.[6] Among these, malignant hypertension (with studies showing a combined incidence as high as 59%–87.5%)[9,10], pregnancy (with studies showing a combined incidence as high as 56%–86%)[11,12], kidney transplantation (with studies showing a combined incidence as high as 40%)[9], hematopoietic stem cell transplantation (with studies showing a combined incidence as high as 10%–80%)[13], and autoimmune diseases[14] require special attention.
The endothelial cells damaged after the patient’s injury lead to endothelial cell activation, pro-coagulant factors, and pro-inflammatory phenotypes that can induce a “second hit” of complement, thus sustaining and exacerbating TMA.[6] Furthermore, a vicious cycle forms between the patient’s genetic susceptibility, triggering factors, and the generation of terminal complement components C5a and C5b9 (MAC), endothelial cell and platelet activation/damage, and thrombin generation, further promoting the onset and development of aHUS.[15]
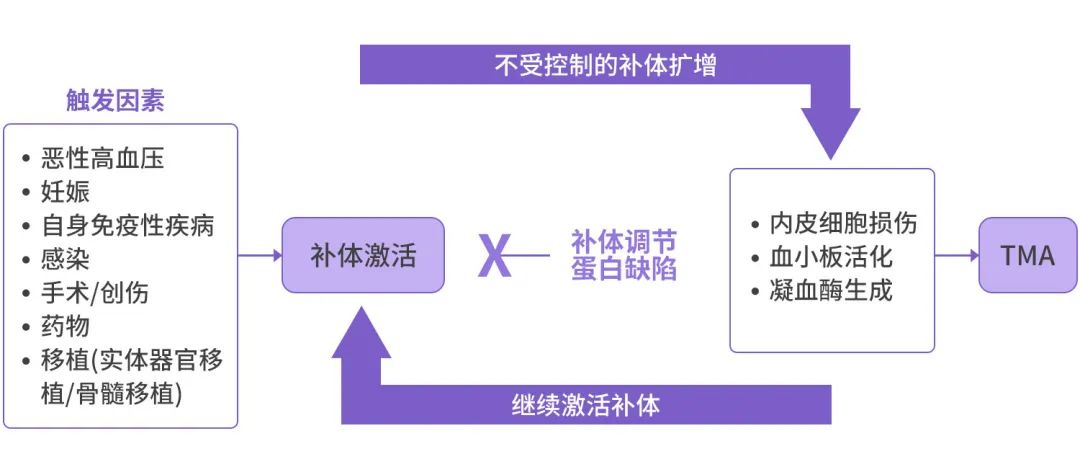
Figure 2: Interaction of Triggering Factors and Complement Activation Promoting the Onset and Development of aHUS[15]
1.3 Clinical Attention is Required for TMA Patients with Concomitant Triggering Factors, and Etiological Investigation is Necessary
Malignant Hypertension: Severe hypertension patients may present systemic signs of TMA, such as thrombocytopenia, microangiopathic hemolytic anemia, or histological signs of TMA in kidney biopsies, therefore, the clinical manifestations and pathology of the two diseases have considerable overlap.[16]
Many aHUS patients have malignant hypertension, but only about 5%–15% of malignant hypertension patients will progress to TMA.[16] The fundamental cause of the latter may be glomerulonephritis rather than hypertension. Therefore, malignant hypertension is both a triggering factor for aHUS and a potential clinical manifestation of the disease, making it sometimes difficult to distinguish causality.
In such cases, the following clinical and histological features may help determine whether the patient’s underlying cause is hypertension rather than primary aHUS: male; age > 45 years; history of hypertension and/or cessation of antihypertensive treatment; rapid (within < 72 hours) resolution of hematological TMA features after strict hypertension control; left ventricular hypertrophy; and no need for dialysis. For patients with TMA features undergoing kidney biopsy, if the results show no glomerular thrombosis, it may assist in excluding complement-mediated thrombotic microangiopathy (CM-TMA).[16]
Pregnancy: Studies show that 79% of pregnancy-related aHUS commonly occurs postpartum.[17] Preeclampsia/HELLP syndrome is common in late pregnancy, and thrombotic thrombocytopenic purpura (TTP) related to ADAMTS13 deficiency mainly occurs in mid to late pregnancy.[18] The vast majority of pregnancy and postpartum TMA can resolve spontaneously after delivery; if TMA signs persist for more than 48 hours after delivery/termination of pregnancy, it strongly suggests the possibility of complement-mediated aHUS.[19]
Kidney Transplantation: Kidney transplantation is also a strong triggering factor for new or recurrent aHUS. Studies show that about 0.8%–14% of kidney transplant patients will experience post-transplant TMA (PT-TMA).[20] Data from the US Renal Data System (USRDS) indicates that the incidence of new PT-TMA is much higher than that of recurrent aHUS (90% vs 10%), but for patients with a history of aHUS, the risk of PT-TMA is much higher than for those without a history (29% vs. 0.8%).[20] This may indicate a recurrence of aHUS. PT-TMA and recurrent aHUS can occur at any time post-transplant, but are mainly concentrated within the first 3 months after transplantation, often accompanied by more complement activation events such as ischemia-reperfusion injury.[20]
Hematopoietic Stem Cell Transplantation (HSCT): Studies show that the incidence of adult HSCT-TMA is about 4%–68%, while that of pediatric HSCT-TMA is about 3%–39%.[21] Multiple factors of complement activation after hematopoietic stem cell transplantation [preconditioning, medications, graft-versus-host disease (GVHD), and infections] can jointly cause endothelial injury, leading to TMA.[21] However, the diagnostic rate of HSCT-TMA is low; statistics show that only half of suspected cases are confirmed.[21] The reason may be a lack of consensus on diagnostic criteria, thus clinical attention is required.
Autoimmune Diseases: Autoimmune diseases such as antiphospholipid syndrome (APS), especially catastrophic antiphospholipid syndrome (CAPS) and systemic lupus erythematosus (SLE) related TMA/aHUS are common.[14] Studies show that complement activation events are found in 85.7% of CAPS patients, 35.6% of APS patients, and 6.8% of SLE patients, with the incidence of complement regulatory gene abnormalities being as high as 60% in CAPS patients, 21.8% in APS, and 28.6% in SLE.[14] Additionally, TMA/aHUS associated with SLE/LN also has multiple influencing factors and is often present with antiphospholipid antibody syndrome, malignant hypertension, infections, etc., thus clinical differential diagnosis is also necessary.[22]
Other Factors: Triggers such as infections, medications, and tumors also require clinical attention; if patients exhibit TMA symptoms, further differentiation is also necessary.
▲ Scroll up and down to view▼
Therefore, clinical attention is particularly needed for TMA patients with triggering factors or concomitant diseases, focusing on whether they may be aHUS patients.
02
Diagnosis of Disease: aHUS is a Diagnosis of Exclusion, Genetic Testing Can Assist Prognosis Assessment
The main clinical manifestations of aHUS are the “triad” of thrombocytopenia, microangiopathic hemolytic anemia, and end-organ (mainly renal) damage.[3]
Currently, there are no specific biomarkers to definitively establish the clinical diagnosis of aHUS, and the pathology of kidney biopsy in aHUS is not specific, making it difficult to make a definitive pathological diagnosis.[23] Therefore, for patients who do exhibit the triad of TMA, clinical practice mainly employs a diagnosis of exclusion.[1]
When diagnosing, first assess the patient’s thrombocytopenia (platelet count < 150 × 109/L or a drop of > 25% from baseline), microangiopathic hemolytic anemia (peripheral blood smear showing > 1% fragmented red blood cells, lactate dehydrogenase elevated > 1.5 times the upper limit of normal reference, combined with decreased haptoglobin and hemoglobin), and accompanying organ damage (kidney, nervous system, cardiovascular, gastrointestinal, respiratory systems) to confirm TMA. Further exclude TTP, STEC-HUS, and ultimately confirm aHUS.[3]
Differential Diagnosis:
Typical hemolytic uremic syndrome: Also known as Shiga toxin-producing Escherichia coli hemolytic uremic syndrome (STEC-HUS), severe diarrhea or bloody stool is the main differentiating point, with stool cultures able to isolate Shiga toxin-producing E. coli or positive anti-endotoxin antibody IgM testing.[23]
Thrombotic thrombocytopenic purpura (TTP): Mainly caused by the loss of ADAMTS13; TTP patients have decreased activity of ADAMTS13, often below 10% of normal values, and serum anti-ADAMTS13 antibodies are positive.[24] When ADAMTS13 results cannot be obtained in a timely manner, the PLASMIC scoring system can be used for assessment.[24]
▲ Scroll up and down to view▼
For TMA patients with triggering factors/concomitant diseases, if the condition continues to progress despite adequate treatment and management of the underlying cause, aHUS should be considered.[1]
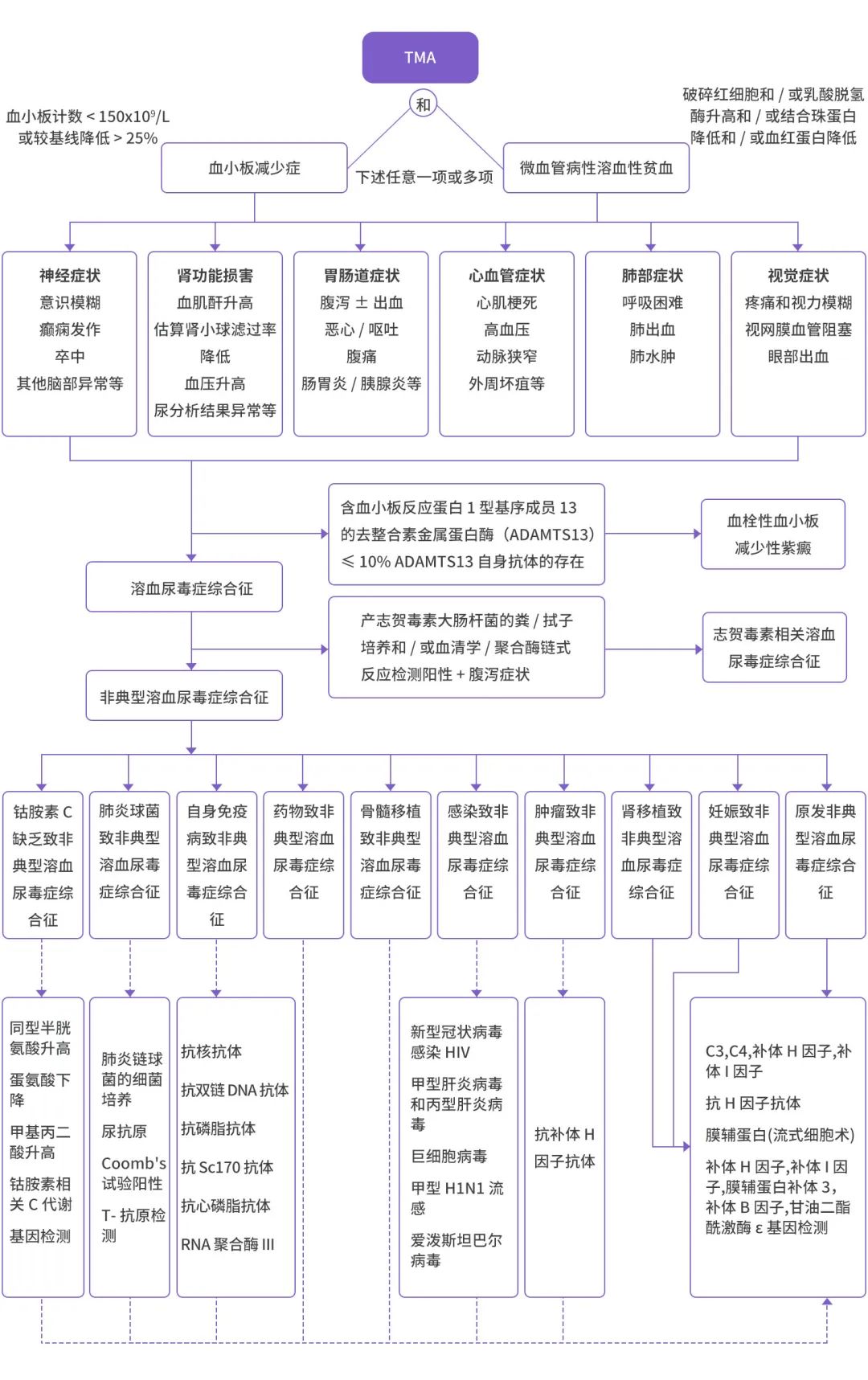
Figure 3: Diagnostic and Differential Diagnosis Flowchart for Patients with Triggering Factors[1,25]
Additionally, when clinical consideration is given to aHUS, various specific protein detection methods can also be used for auxiliary diagnosis: quantitative complement components and their regulatory factors (CFH, C3, CFI, MCP, C4, and CFB), complement activity markers (CH50 and AH50), screening for anti-CFH autoantibodies; genetic screening for known genes that cause aHUS (CFH, CFI, MCP, C3, CFB, THBD, DGKE, and CFHR).[23] Although genetic complement testing is not essential for diagnosis, it can assess prognosis and guide long-term treatment plans.
03
Treatment of Disease: Guidelines Recommend Complement Inhibitor Therapy
The treatment strategy for aHUS is to correct the dysregulation of the complement system. Prior to the use of complement inhibitors, plasma therapy was the first-line choice, but plasma therapy is ineffective for patients with complement gene abnormalities and does not address high recurrence rates and progression of renal damage.[3]
At the 2024 European Renal Association (ERA) Annual Meeting, reports also indicated that aHUS patients undergoing plasma exchange treatment had poor renal prognosis.[26]
With the emergence of complement inhibitors and the increasing evidence base, guidelines and consensus from both domestic and international sources now recommend complement inhibitor therapy for aHUS patients.[1,3,27-28]
The Kidney Disease: Improving Global Outcomes (KDIGO) “Expert Consensus on the Treatment of Atypical Hemolytic Uremic Syndrome and C3 Glomerulopathy” indicates that all clinically diagnosed primary aHUS patients are suitable for complement inhibitor therapy.[25]
The 2017 “Expert Statement on the Standard of Care for Critically Ill Adult Patients with Atypical Hemolytic Uremic Syndrome” recommends that for ICU patients suspected of aHUS, plasma therapy should begin within 4-8 hours of admission or diagnosis of TMA; for patients with first-time TMA, once aHUS is diagnosed (ADAMTS13 > 10% and STEC negative), treatment should be immediately switched to C5 inhibitors.[27]
China’s “Expert Consensus on the Diagnosis and Treatment of Complement-Related Kidney Diseases” (2024 edition) also recommends that the treatment goal for aHUS is to achieve TMA remission as quickly as possible, with no early recurrence. Patients diagnosed with aHUS should receive complement inhibitor therapy as soon as possible.[1] After confirming complement-mediated aHUS, efforts should be made to initiate eculizumab treatment within 24-48 hours of onset to control disease progression.
▲ Scroll up and down to view▼
As of now, the C5 complement inhibitor eculizumab has been approved in China for aHUS indications, while other complement inhibitors such as iptacopan and ravulizumab have been approved for paroxysmal nocturnal hemoglobinuria (PNH) indications in China, and global multicenter clinical studies regarding aHUS [iptacopan (NCT04889430), ravulizumab (NCT04861259, NCT04858265)] are still ongoing.[1,29]
A retrospective analysis included 29 aHUS patients with triggering factors from 11 renal centers in Spain (15 drug-related, 8 systemic disease-related, 2 postpartum, 2 cancer-related, 1 related to acute fluid rejection, and 1 related to intestinal lymphangiectasia), all of whom received eculizumab treatment. The primary endpoint of the study was the resolution of TMA symptoms, defined as platelet count (> 150 × 109/L) and normalization of hemoglobin, disappearance of all markers of MAHA, and improvement of renal function (≥ 25% decrease in serum creatinine from the start of eculizumab administration). The results showed that among patients receiving eculizumab treatment, 69% (20/29) had rapid resolution of TMA symptoms; 71.4% (10/14) were able to stop dialysis; 51.7% (15/29) had a serum creatinine decrease of ≥ 50%, and 35% (10/29) had an eGFR ≥ 60 mL/min/1.73 m2 at the last follow-up.[30]
Furthermore, the KDIGO consensus also points out that early initiation of C5 complement inhibitors can help patients recover kidney function, and continued use of C5 inhibitors for more than 26 weeks may provide some patients the possibility of stopping dialysis.[25] At the 2024 ERA meeting, studies also reported that for patients already on dialysis, continuing C5 inhibitor treatment for over 26 weeks may also provide some patients the possibility of stopping dialysis.[31]
The KDIGO “Expert Consensus on the Treatment of Atypical Hemolytic Uremic Syndrome and C3 Glomerulopathy”[25] states that treatment for all complement-related aHUS patients should last at least 6-12 months, and consideration to stop complement inhibitor therapy may be made based on the patient’s specific circumstances (genetic mutations, history of transplantation, history of recurrence, history of dialysis, etc.) only after renal function has returned to baseline (or stabilized in cases of chronic kidney disease) for at least 3 months. The risk of recurrence after cessation of treatment should also be comprehensively assessed.
04
Conclusion
aHUS is a rare complement-mediated thrombotic microangiopathy (TMA) that is usually quite critical, and its onset depends on the interaction between the patient’s genetic predisposition and environmental triggering factors.
The presence of multiple triggering factors makes the clinical diagnosis of aHUS more challenging; therefore, clinicians should pay close attention to such patients, focusing on the investigation of whether they may be aHUS patients. For aHUS patients with concomitant triggering factors, adequate treatment of the underlying causes should be provided, and timely initiation of complement inhibitor therapy should be ensured.
This material is provided by AstraZeneca to meet your medical information needs and is intended for reference by healthcare professionals only, not for promotional purposes.
Approval No.: CN-141386, Expiry Date: 2025-08-18
References
[1]. Expert Group of the Department of Nephrology, Peking University Health Science Center. Expert Consensus on the Diagnosis and Treatment of Complement-Related Kidney Diseases [J]. Chinese Journal of Internal Medicine, 2024, 63(03): 258-271.
[2]. Noris M, Caprioli J, Bresin E, et al. Relative role of genetic complement abnormalities in sporadic and familial aHUS and their impact on clinical phenotype. Clin J Am Soc Nephrol. 2010;5(10):1844-1859.
[3]. China Rare Disease Alliance Children’s Atypical Hemolytic Uremic Syndrome Expert Committee, National Children’s Medical Center (Beijing Children’s Hospital affiliated to Capital Medical University), Editorial Board of Chinese Practical Pediatrics. Expert Consensus on the Diagnosis and Treatment of Children’s Atypical Hemolytic Uremic Syndrome (2023 Edition) [J]. Chinese Journal of Practical Pediatrics, 2023, 38(6): 401-412.
[4]. Meri S. Complement activation in diseases presenting with thrombotic microangiopathy. Eur J Intern Med. 2013;24(6):496-502.
[5]. Raina R, Vijayvargiya N, Khooblall A, et al. Pediatric Atypical Hemolytic Uremic Syndrome Advances. Cells. 2021;10(12):3580.
[6]. Praga M, Rodríguez de Córdoba S. Secondary atypical hemolytic uremic syndromes in the era of complement blockade. Kidney Int. 2019;95(6):1298-1300.
[7]. Harris CL, Pouw RB, Kavanagh D, Sun R, Ricklin D. Developments in anti-complement therapy; from disease to clinical trial. Mol Immunol. 2018;102:89-119.
[8]. Brocklebank V, Wood KM, Kavanagh D. Thrombotic Microangiopathy and the Kidney. Clin J Am Soc Nephrol. 2018;13(2):300-317.
[9]. Timmermans SAMEG, Damoiseaux JGMC, Werion A, Reutelingsperger CP, Morelle J, van Paassen P. Functional and Genetic Landscape of Complement Dysregulation Along the Spectrum of Thrombotic Microangiopathy and its Potential Implications on Clinical Outcomes. Kidney Int Rep. 2021;6(4):1099-1109.
[10]. Timmermans SAMEG, Abdul-Hamid MA, Potjewijd J, et al. C5b9 Formation on Endothelial Cells Reflects Complement Defects among Patients with Renal Thrombotic Microangiopathy and Severe Hypertension. J Am Soc Nephrol. 2018;29(8):2234-2243.
[11]. Fakhouri F, Roumenina L, Provot F, et al. Pregnancy-associated hemolytic uremic syndrome revisited in the era of complement gene mutations. J Am Soc Nephrol. 2010;21(5):859-867.
[12]. Bruel A, Kavanagh D, Noris M, et al. Hemolytic Uremic Syndrome in Pregnancy and Postpartum. Clin J Am Soc Nephrol. 2017;12(8):1237-1247.
[13]. Ardissino G, Capone V, Tedeschi S, Porcaro L, Cugno M. Complement System as a New Target for Hematopoietic Stem Cell Transplantation-Related Thrombotic Microangiopathy. Pharmaceuticals (Basel). 2022;15(7):845.
[14]. Chaturvedi S, Braunstein EM, Yuan X, et al. Complement activity and complement regulatory gene mutations are associated with thrombosis in APS and CAPS. Blood. 2020;135(4):239-251.
[15]. Laurence J, Haller H, Mannucci PM, Nangaku M, Praga M, Rodriguez de Cordoba S. Atypical hemolytic uremic syndrome (aHUS): essential aspects of an accurate diagnosis. Clin Adv Hematol Oncol. 2016;14 Suppl 11(11):2-15.
[16]. Wehrmann F, von Bergwelt-Baildon A, Schönermarck U. Severe hypertension and (renal) thrombotic microangiopathy: solving the puzzle. J Nephrol. 2023;36(8):2175-2177.
[17]. Fakhouri F, Roumenina L, Provot F, et al. Pregnancy-associated hemolytic uremic syndrome revisited in the era of complement gene mutations. J Am Soc Nephrol. 2010;21(5):859-867.
[18]. Smith P, Abdelmaguid A, Clark K, Bramham K. Lessons for the clinical nephrologist: recurrent pregnancy-associated thrombotic microangiopathy (TMA) with a known complement mutation and features of preeclampsia; a diagnostic and therapeutic dilemma. J Nephrol. 2021;34(5):1673-1676.
[19]. Gupta M, Feinberg BB, Burwick RM. Thrombotic microangiopathies of pregnancy: Differential diagnosis. Pregnancy Hypertens. 2018;12:29-34.
[20]. Ávila A, Gavela E, Sancho A. Thrombotic Microangiopathy After Kidney Transplantation: An Underdiagnosed and Potentially Reversible Entity. Front Med (Lausanne). 2021;8:642864.
[21]. Mahmoudjafari Z, Alencar MC, Alexander MD, Johnson DJ, Yeh J, Evans MD. Hematopoietic stem cell transplantation-associated thrombotic microangiopathy and the role of advanced practice providers and pharmacists. Bone Marrow Transplant. 2023;58(6):625-634.
[22]. Yang Xinglin, Zhang Shangzhu, Xu Dong, et al. Systemic lupus erythematosus complicated by thrombotic microangiopathy [J]. Chinese Journal of Clinical Immunology and Allergy, 2018, 12(05): 545-551.
[23]. Dai Yiping, Ruan Yiping, Hong Fuyuan. Research Progress on the Treatment of Atypical Hemolytic Uremic Syndrome. Journal of Rare Diseases. 2022(07): 1-4.
[24]. Dai Yanling, Dai Siyuan, Li Xiaozhao, et al. Clinical Diagnosis and Treatment Progress of Thrombotic Microangiopathy [J]. Chinese Medical Journal, 2018, 98(48): 3987-3990.
[25]. Goodship TH, Cook HT, Fakhouri F, et al. Atypical hemolytic uremic syndrome and C3 glomerulopathy: conclusions from a “Kidney Disease: Improving Global Outcomes” (KDIGO) Controversies Conference. Kidney Int. 2017;91(3):539-551.
[26]. Lamis Khedr, Shaimaa Zaki and Reem Elsharabasy. Renal outcomes of postpartum atypical hemolytic uremic syndrome treated with plasma exchange. 2024 ERA. Abstract#2262.
[27]. Azoulay E, Knoebl P, Garnacho-Montero J, et al. Expert Statements on the Standard of Care in Critically Ill Adult Patients With Atypical Hemolytic Uremic Syndrome. Chest. 2017;152(2):424-434.
[28]. Ávila A, Cao M, Espinosa M, Manrique J, Morales E. Recommendations for the individualised management of atypical hemolytic uremic syndrome in adults. Front Med (Lausanne). 2023;10:1264310.
[29]. Antonucci L, Thurman JM, Vivarelli M. Complement inhibitors in pediatric kidney diseases: new therapeutic opportunities. Pediatr Nephrol. 2024;39(5):1387-1404.
[30]. Cavero T, Rabasco C, López A, et al. Eculizumab in secondary atypical haemolytic uraemic syndrome. Nephrol Dial Transplant. 2017;32(3):466-474.
[31]. Shuichi Ito, Masanori Matsumoto, Akihiko Shimono, Hirofumi Teranishi, Shoichi Maruyama. Characteristics of patients with aHUS who discontinued dialysis after eculizumab treatment: sub-analysis of post-marketing surveillance in Japan. 2024 ERA. Abstract#550
Yimaitong is a professional online doctor platform, “perceiving the world’s medical pulse, assisting clinical decision-making in China” is the platform’s mission. Yimaitong has a series of products such as “Clinical Guidelines”, “Medication Reference”, “Medical Literature King”, “Medical Knowledge Source”, “e-Research”, “e-Pulse Broadcast”, etc., comprehensively meeting the needs of medical workers for clinical decision-making, acquiring new knowledge, and improving research efficiency. For HCP viewing only.
