Disclaimer: This article aims to share learning records and may contain errors. Although the specific images and texts are cited, the copyright belongs to the publisher and the original authors; if there is any infringement, please contact us to delete the relevant content within 30 days of publication.
1.5 Intramolecular C(sp3)-Hydrogen Bond Insertion
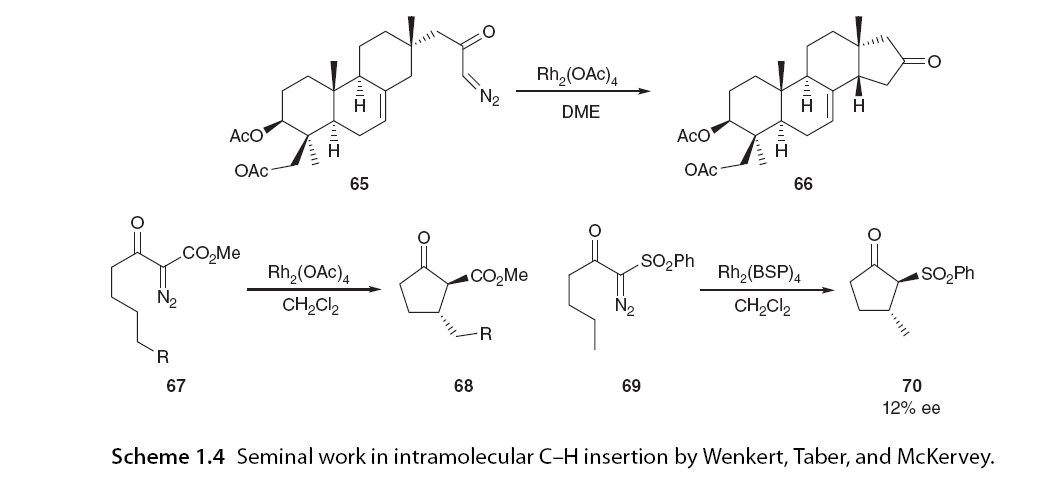
Intramolecular C-H insertion was not considered an effective transformation until Wenkert et al. [10] and Taber and Petty [11] independently reported rhodium-catalyzed α-diazo ketone cyclization in the early 1980s, at ages 65 and 67, respectively. Prior to this work, the application of tetracarboxylic acid dirhodium in intermolecular C-H insertion by Teyssié allowed intramolecular C-H insertion to become a powerful synthetic route for constructing five-membered rings. Taber recognized the potential for asymmetric induction in this transformation by introducing chirality in the α-diazo ketone precursor’s auxiliary [57], followed by McKervey and colleagues who first reported the application of chiral dirhodium catalysts in asymmetric reactions involving intramolecular C-H insertion, providing moderate yields of 12% ee for cyclopentanone (Scheme 1.4). Since these pioneering reports, significant progress has been made in the field of metal-catalyzed asymmetric intramolecular C-H insertion, establishing it as a reliable method for C-C bond formation. For clarity, this chapter will be organized based on chemical, regio-, stereo-, and enantiomeric selectivity, focusing on the literature surrounding rhodium-catalyzed C-H insertion, which dominates the field.
1.5.1 Chemical Selectivity
1.5.1.1 Catalyst Effects
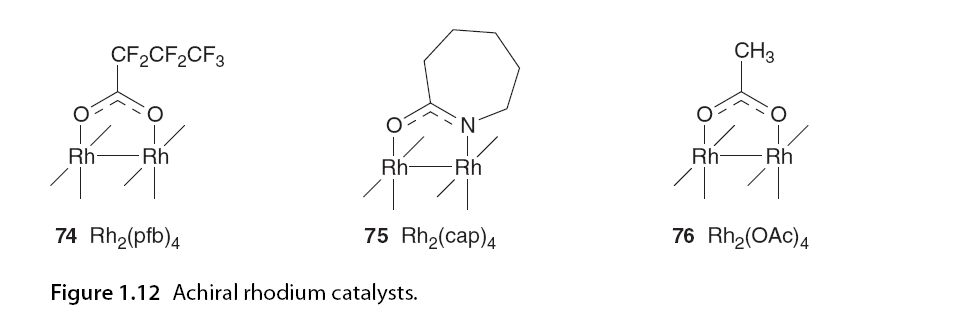
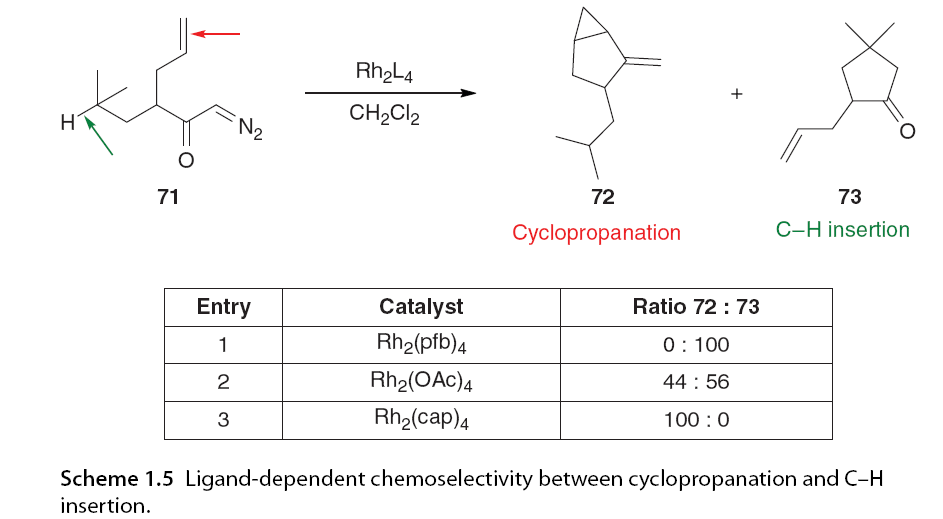
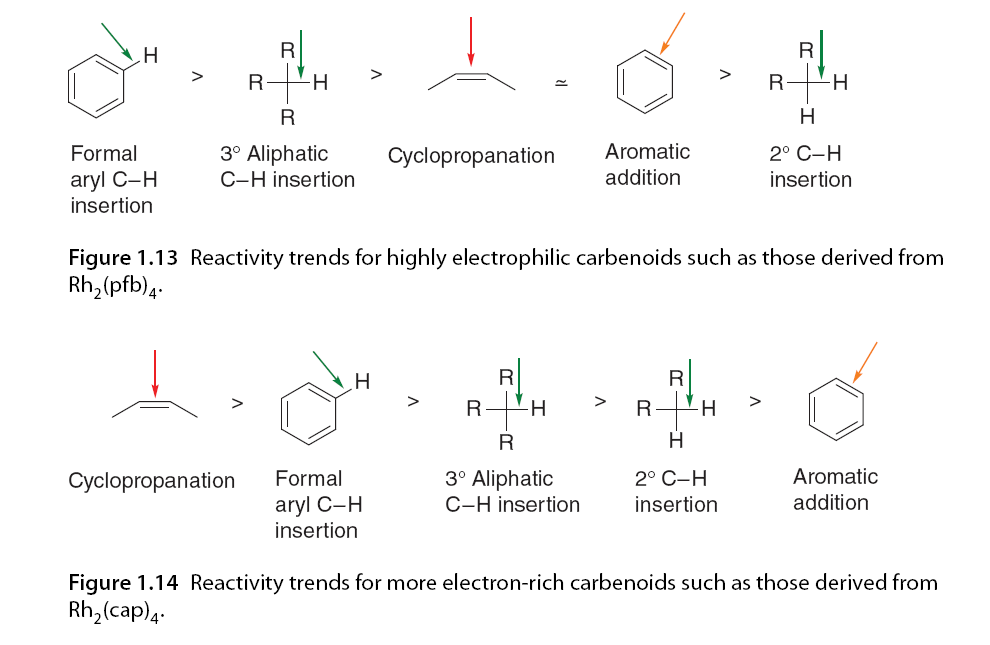
Carbene compounds are more prone to C-H insertion than other carbene types, which has drawn significant interest from synthetic chemists. In rhodium-catalyzed carbene-mediated transformations, the selectivity of C-H insertion was found to depend on the catalyst, particularly the electronic properties of the ligands. Padwa, Doyle, and colleagues conducted a series of intramolecular competition experiments [58,59]. α-Diazo ketone 71 has the potential for cyclopropanation or C(sp3)-H insertion; it was observed that C-H insertion was the only reaction pathway when using the highly electrophilic carbene derived from Rh2(pfb)4 (74). In contrast, the carbene derived from Rh2(cap)4 (75), which has higher electron content, led to a shift in chemical selectivity, favoring cyclopropanation entirely. The electronic properties of Rh2(OAc)4 (76) were intermediate between the other two catalysts, showing little chemical selectivity (Figure 1.12 and Scheme 1.5).
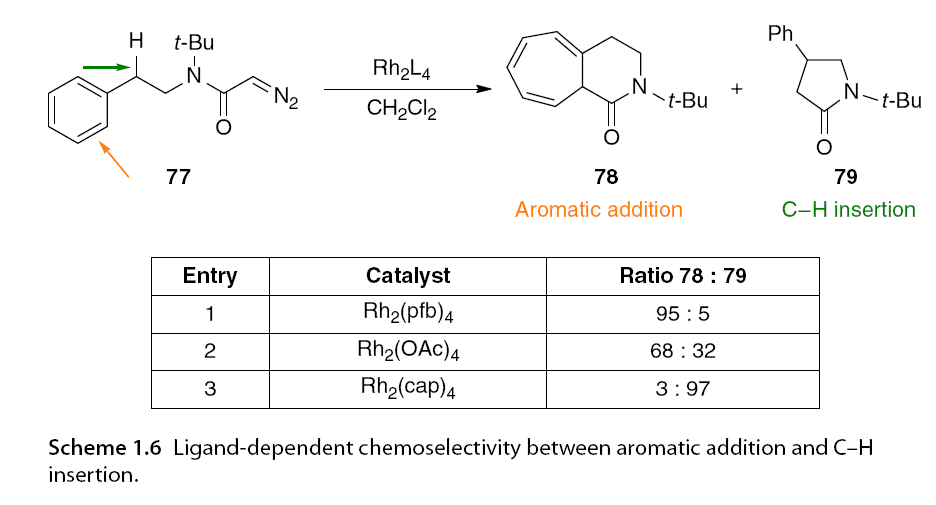
Another example of ligand-dependent chemical selectivity was observed with α-diazoacetamide 77, which underwent rhodium-catalyzed C-H insertion and aromatic addition reactions at the insertion site. In this experiment, C-H insertion was the preferred reaction pathway when using Rh2(cap)4, while Rh2(pfb)4 predominantly produced aromatic addition products. No insertion was observed in the tert-butyl control group (Scheme 1.6). Although only two examples are shown here, a series of extensive competition experiments highlighted prominent reactivity trends based on catalyst selection.
1.5.1.2 Substrate Effects
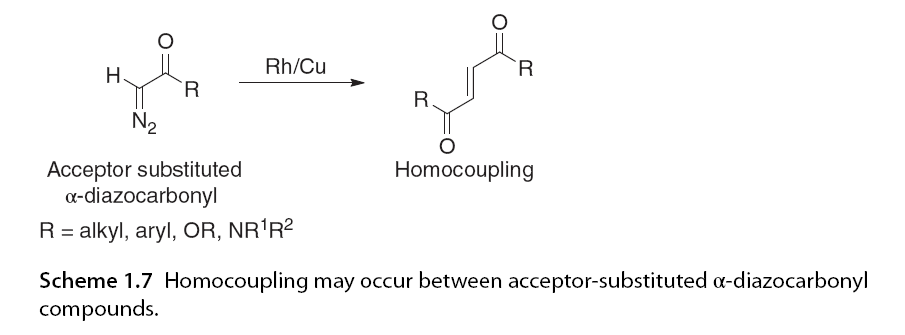
Another factor influencing C-H insertion selectivity is the substitution pattern of the α-diazo carbonyl substrate. Typically, compounds with electron-withdrawing groups (EWG) adjacent to the diazo part lead to more reactive carbene intermediates. This can make homodimerization a competing side reaction. Such homodimerization is less favorable for substrates with lower reactivity, or donor/acceptor α-diazo carbonyl compounds (Scheme 1.7).
Interestingly, Padwa and Moody showed that if a hydroxyl group is present in the substrate, O-H insertion will dominate over any other carbene-mediated transformations, regardless of the dirhodium catalyst used (Scheme 1.8). Although yields varied, O-H insertion was the only product obtained in the absence of cyclopropanation, aromatic addition, or aliphatic evidence of aromatic C-H insertion [61].
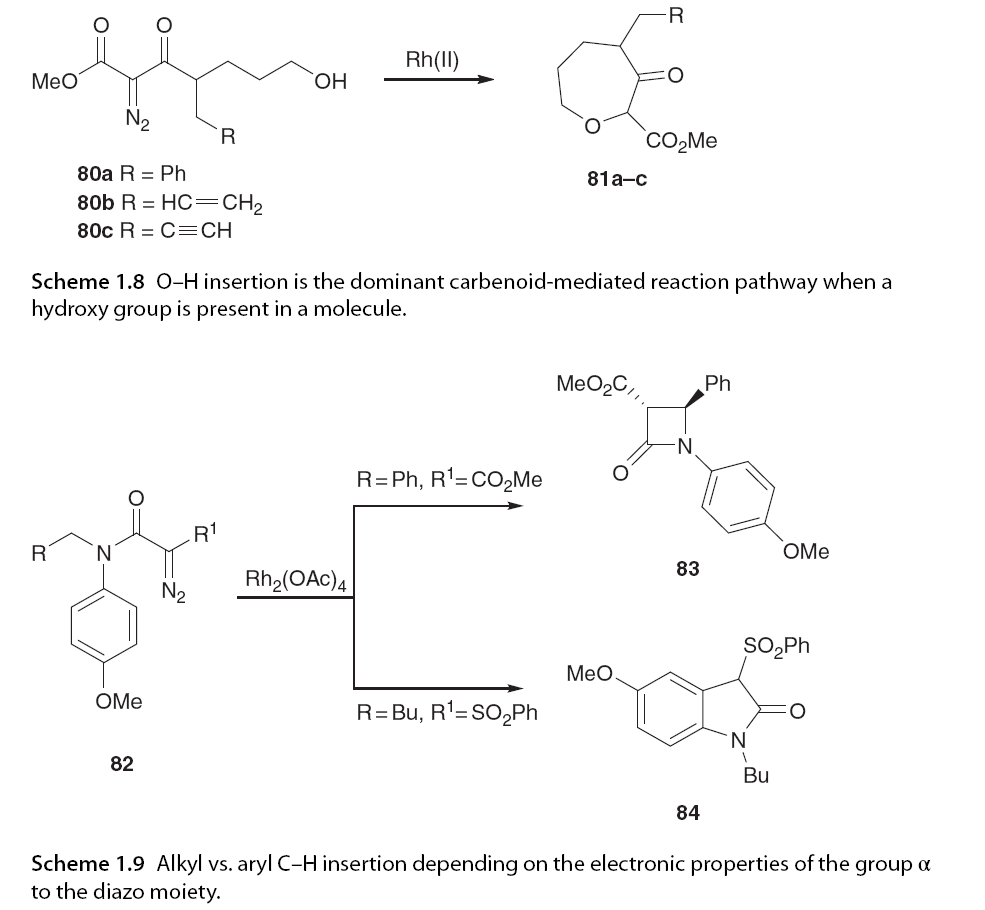
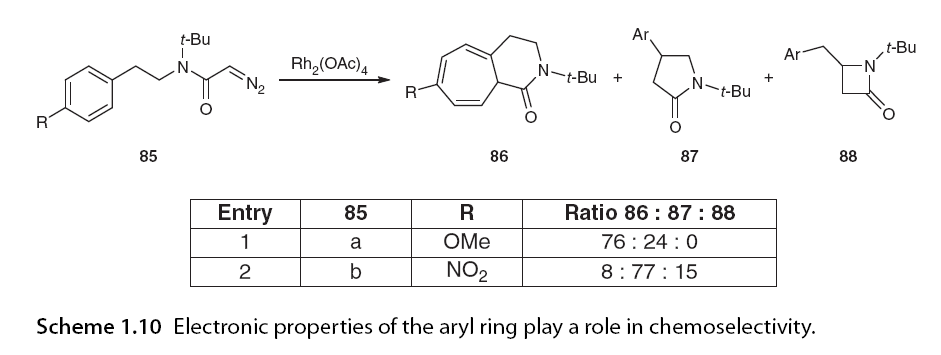
Wee and colleagues designed a study consisting of 11 α-diazo anilines 82, altering the N-substituents to investigate the C-H insertion of alkyl versus aryl (Scheme 1.9). In all cases, alkyl C-H insertion was the only pathway observed (e.g., 83). In contrast, when the α-diazo ester part was modified to phenyl sulfone, only formal aromatic C-H insertion was observed (one example, 84). This can be rationalized by the stronger electrophilicity of the acetyl or phenyl sulfonyl groups leading to more electrophilic carbene compared to ester-derived carbene [62]. It should be noted that while this reaction is strictly referred to as “aromatic C-H insertion,” it should not be, as the mechanism is aromatic substitution rather than C-H insertion. The study also indicated that the electronic properties of the aromatic ring play a significant role, with strong electron-withdrawing nitro substituents disfavoring aromatic addition and promoting C-H insertion as the primary reaction pathway (Scheme 1.10) [59].
1.5.2 Regioselectivity
According to Taber’s investigation [11,12,63], it can be established that intramolecular C-H insertion preferentially forms five-membered cyclic compounds. This fact has been utilized to construct five-membered carbonyl heterocyclic systems such as cyclopentanones, dihydrofurans, γ-lactones, and γ-lactams. Although 1,5 C-H insertion is favored due to entropy, steric or electronic factors can override this preference, leading to the formation of larger or smaller ring sizes. Generally, if a C-H bond is formed, 1,4 C-H insertion can occur, activated by adjacent heteroatoms (usually nitrogen or oxygen), while 1,6 C-H insertion has been observed for more rigid systems. Even 1,3 β-toluenesulfonyl α-diazo carbonyl compounds have been reported to undergo C-H insertion.
1.5.2.1 Formation of Three-Membered Rings
While three-membered rings can be constructed through geometrical C-H insertion to form rigid structures, one example of three-membered ring formation has been reported in freely rotating systems. Wang and colleagues found that α-diazo carbonyl compounds with β-toluenesulfonyl functional groups exhibited excellent enantiomeric selectivity in cyclopropane formation under oC insertion (Scheme 1.11) [64].
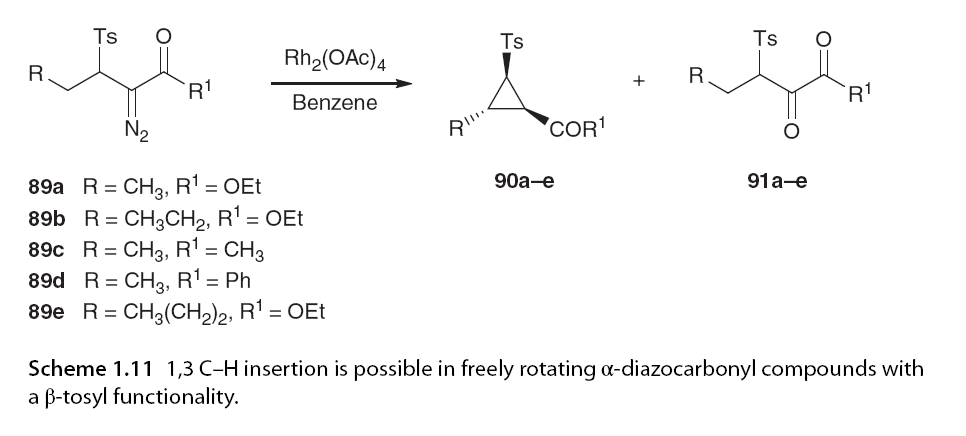
When the reaction was conducted under strictly anaerobic conditions to prevent the formation of oxidation products 91, 1,3 C-H insertion was the major reaction pathway, with the exception of one case where the remaining pathways were observed. Interestingly, α-diazo carbonyl compound 89e has the potential to undergo either 1,3 C-H insertion or 1,5 C-H insertion. Using Rh2(OAc)4 as the catalyst, the distribution of insertion products was uniform. However, if the catalyst had more electron-withdrawing ligands, such as Rh2(tfa)4, 1,3 C-H insertion was favored. This study indicates the significant impact of adjacent groups and catalyst selection on regioselectivity. At the time of writing, 1,3 C-H insertion has only been reported with non-cardiac catalysts.
1.5.2.2 Formation of Four-Membered Rings
The most common four-membered rings formed through C-H insertion are β-lactones and β-lactams. The selective synthesis of these structural motifs is significant due to their presence in many bioactive compounds. The primary obstacle to C-H insertion forming four-membered rings is that when both insertion sites are available, the reaction tends to favor 1,5 C-H insertion. Therefore, it is necessary to restrict access to the δ-C-H insertion site either electronically or spatially to force 1,4-insertion. Attempts to synthesize β-lactones have been challenging, as γ-lactone C-H insertion reactions have almost always been the only reaction products from α-diazo acetate. Doyle reported a notable exception to this trend when he demonstrated the cyclization of alkyl phenyl diazoacetates to yield β-lactones as the major reaction products (Scheme 1.12). In the example shown here, Rh2(S-DOSP)4 was used to produce β-lactones 93 and 95 in high yields, with only trace amounts of γ-lactone products detected, exhibiting moderate enantiomeric selectivity [65].
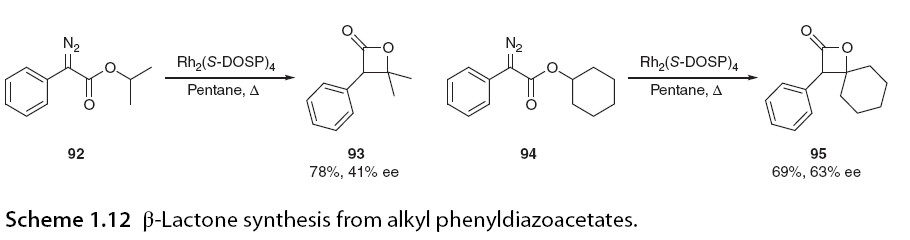
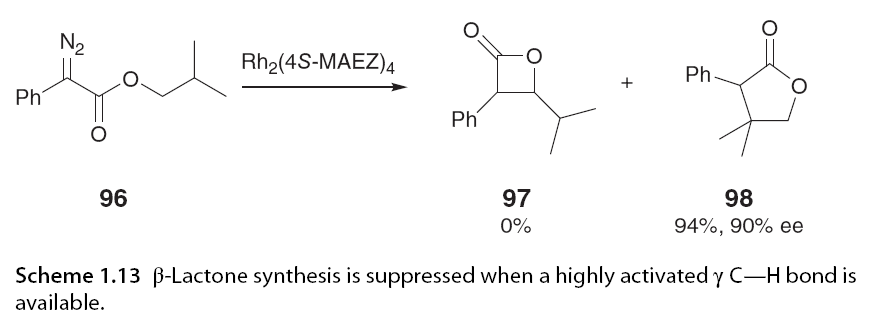
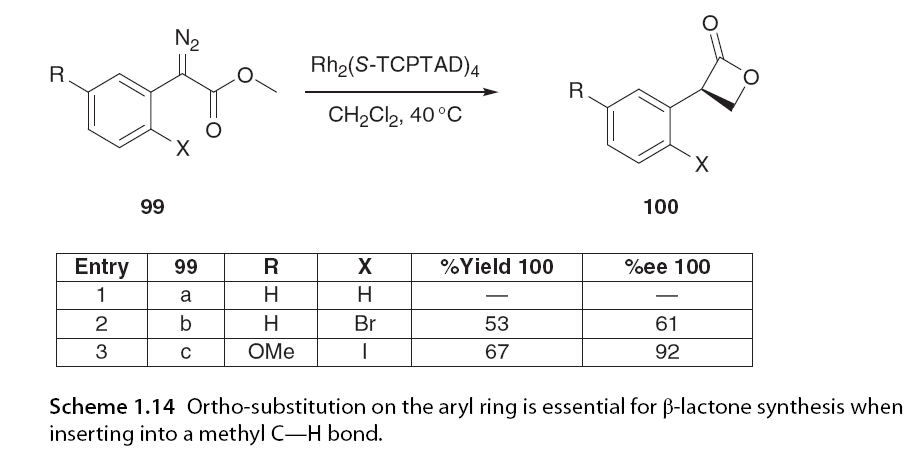
When the γ C-H bond is highly activated, this regioselectivity can be suppressed to allow for insertion. In Scheme 1.13, β-lactones are not formed; instead, γ-lactone 98 is formed with a 94% yield and excellent enantiomeric selectivity. Davis and his colleagues [46] and Bach further expanded this work, demonstrating that not only does substitution at the ester site affect regioselectivity, but substitution on the α-aromatic ring also influences it. The research group observed that ortho-substituents on aryl diazoacetates promoted 1,4-insertion, while Davies reported that ortho-substituents enhanced enantiomeric selectivity when performing methyl C-H insertion in the presence of a methoxy group on the aryl ring (Scheme 1.14).
β-Lactams can be considered one of the most important heterocycles in pharmaceuticals due to their presence in essential broad-spectrum antibiotics such as penicillin and cephalosporins. Intramolecular C-H insertion of α-diazoacetamides can be used to generate β-lactams by activating adjacent C-H bonds through carbene insertion. The first report of β-lactam formation via C-H insertion came from Corey and Felix [67], but the first report of enantioselective β-lactam synthesis via C-H insertion was published by Doyle et al. [68]. In this report, the formation of β-lactams depended on the substituents on the N-alkyl chain of the α-diazoacetamide precursor, with yields of any chiral compound not exceeding 25% in the case of 103a, using the catalyst and achieving moderate enantiomeric selectivity of up to 80% ee with Rh2(4S-MEOX)4 (Scheme 1.15).
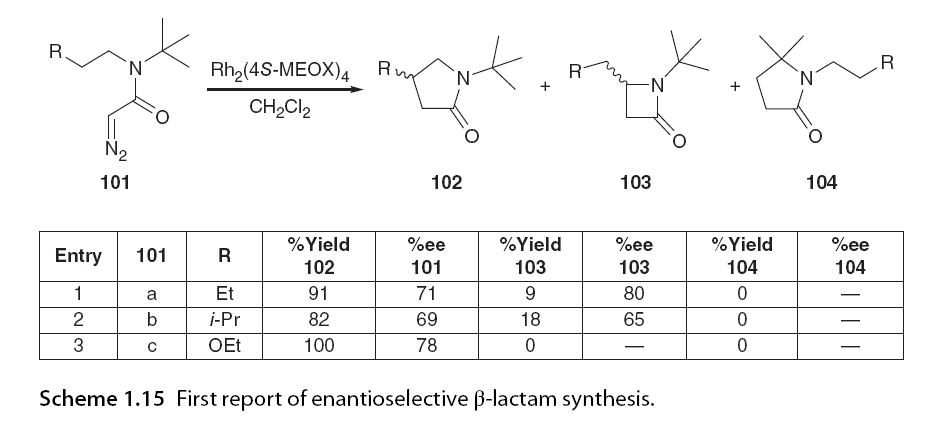
In the preferentially freely rotating alkyl diazoacetamides undergoing 1,5 C-H insertion, Doyle investigated the enantioselective synthesis of β-lactams using more conformationally constrained α-diazo acetamides. In contrast to unsuccessful reports with pyrrolidine, piperidine, or morpholine derivatives, excellent regioselectivity was demonstrated with α-diazo acetamides, azabicycloheptanes, and azabicyclooctanes, achieving >99:1 β:γ-lactam formation, excellent yields, and very high enantiomeric selectivity (Scheme 1.16) [69]. More chiral examples of β-lactams generated via C-H insertion exist, but they all exhibit a high dependence on the substitution patterns of α-diazoacetamides and catalysts [70].
1.5.2.3 Formation of Five-Membered Rings
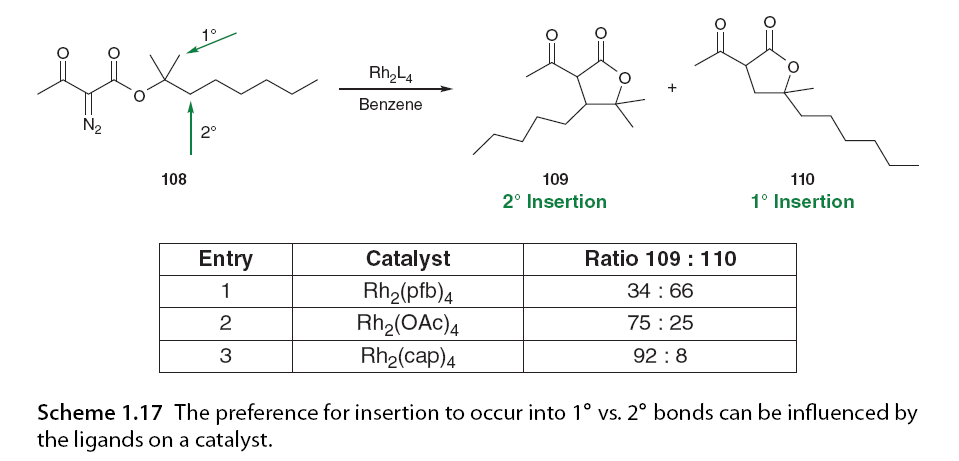
Early studies by Taber and his colleagues [11,57,63] and Doyle et al. [71] have demonstrated that carbene can insert into secondary C-H bonds, while primary C-H insertion is the least favorable. This is due to the accumulation of positive charge at the carbon where insertion occurs; therefore, substitution patterns that can better stabilize the positive charge will promote the insertion process (Scheme 1.17). This selectivity can also be influenced by the electronic properties of the catalyst [19].
1.5.2.4 Formation of Six-Membered Rings
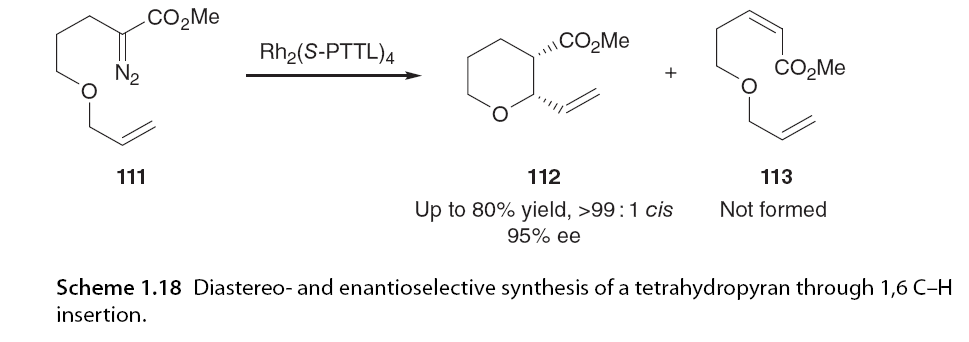
Formation of six-membered rings via C-H insertion has been shown to be strongly dependent on the nature of the α-diazo carbonyl precursor. It has been discovered that by activating the C-H insertion site with adjacent heteroatoms, six-membered rings can be constructed. Extending McKervey’s early work, Ye [72] and Hashimoto and colleagues reported the enantioselective synthesis of six-membered oxygen-containing heterocycles through 1,6 C-H insertion [73]. Using Rh2(S-PTTL)4, tetrahydropyran 112 was synthesized with up to 95% ee and complete cis diastereoselectivity (Scheme 1.18).
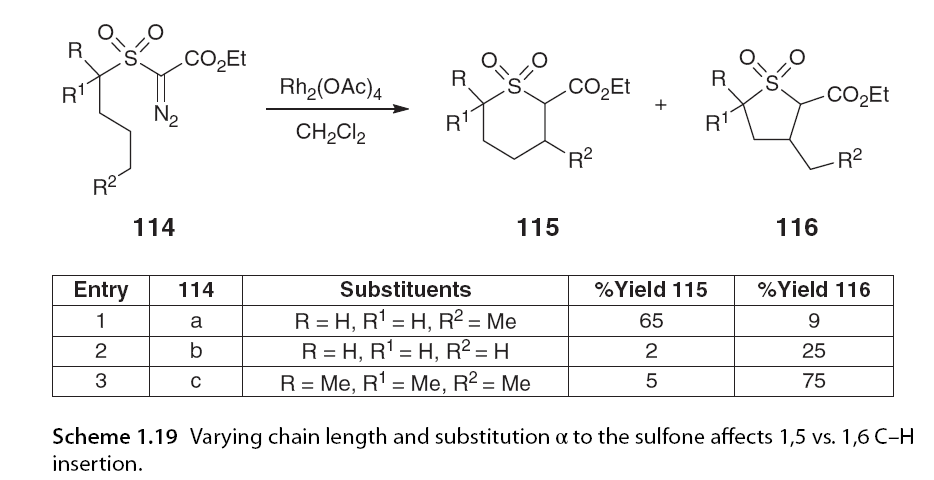
When the reaction was conducted in ether at -60°C, no evidence of competitive β-hydride elimination products 113 was observed. The activation of α-heteroatoms is not always necessary to induce 1,6 C-H insertion, as demonstrated by Novikov and colleagues [74,75], Taber et al. [76], and Du-Bois and colleagues [77] in the synthesis of six-membered sulfoxides and cyclohexanones. Novikov and Taber investigated the effects of different substitutions on regioselectivity patterns on α-diazo carbonyl substrates. Novikov and colleagues examined the substitution patterns on the carbon adjacent to the sulfone and at the insertion site. α-Substituted sulfones favored 1,5 C-H insertion, while longer chain lengths promoted the formation of thiofuran via 1,6 C-H insertion (Scheme 1.19) [75]. Du-Bois and colleagues (Scheme 1.20).
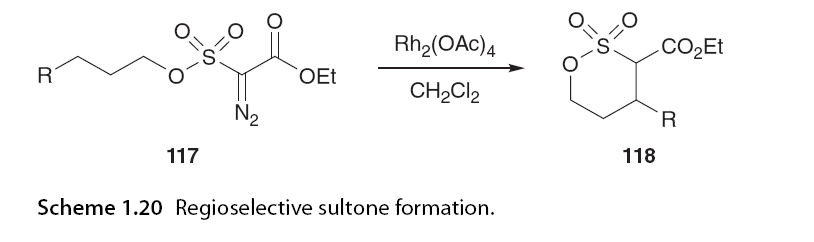
The strong preference for 1,6 C-H insertion is attributed to the absence of cyclic sulfonates and cyclic sulfonyl esters. The formation of five-membered γ-sulfonyl lactones is suppressed as 1,5 C-H insertion requires unfavorable C-S-O bond distortions [77]. The Taber group synthesized cyclohexanones from α-diazo-β-aryl compounds through selective electron-withdrawing aryl substitutions at the diazo part, achieving excellent regioselectivity. The formation of six-membered rings stands in stark contrast to Taber’s pioneering work in the field, where he illustrated how the electronic donor/acceptor nature of carbene affects the selectivity of C-H bonds derived from α-diazo-β-keto esters. Cyclohexanone 120 was synthesized with a selectivity of 99:1, superior to the corresponding cyclopentanone 121 (Scheme 1.21). Du-Bois and colleagues also reported that the stereoselective formation of cyclohexanones is a key step in their synthesis of pufferfish toxins. The dirhodium catalyst used for this transformation was Rh2(HNCOCPh3)4.
[78].
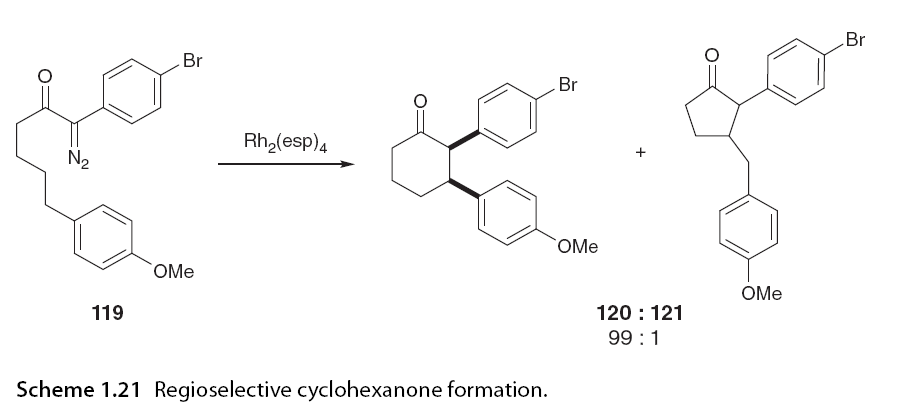
Maguire and colleagues have demonstrated the regioselective synthesis of cis compounds of thiofuran 124 with excellent enantiomeric selectivity [30]. Interestingly, this transformation was achieved using a chiral copper bisoxazoline catalyst complex. Surprisingly, when applying rhodium catalysts to similar substrates 122a, the maximum asymmetric induction achieved was 50% ee using Rh2(S-PTTL)4. When the ester part was replaced with a menthol chiral auxiliary (Scheme 1.22) [79].
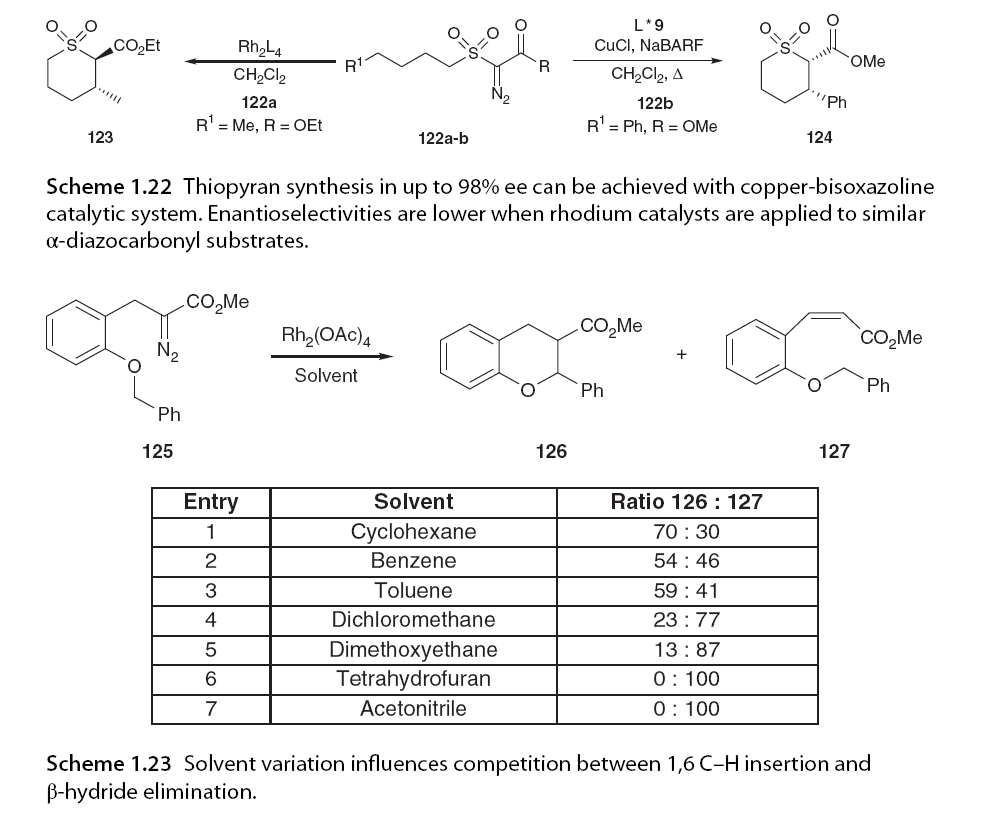
The reaction solvent has also been shown to influence 1,6 C-H insertion and β-hydride elimination. It has been reported that polar solvents such as cyclohexane shift the reaction pathway towards 1,6 C-H insertion, while acetonitrile and tetrahydrofuran favor the β-hydride elimination process entirely (Scheme 1.23) [80].
1.5.3 Discrete Selectivity
1.5.3.1 Substrate Effects
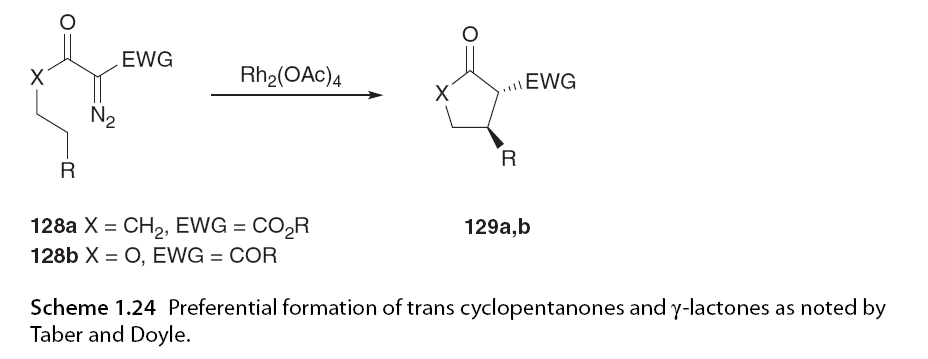
Taber and Ruckle [63,81] and Doyle et al. [71] conducted early studies on the catalytic cyclization reactions of Rh2(OAc)4 with disubstituted α-diazo carbonyl compounds, preferentially forming trans cyclopentanones and γ-lactones (Scheme 1.24).
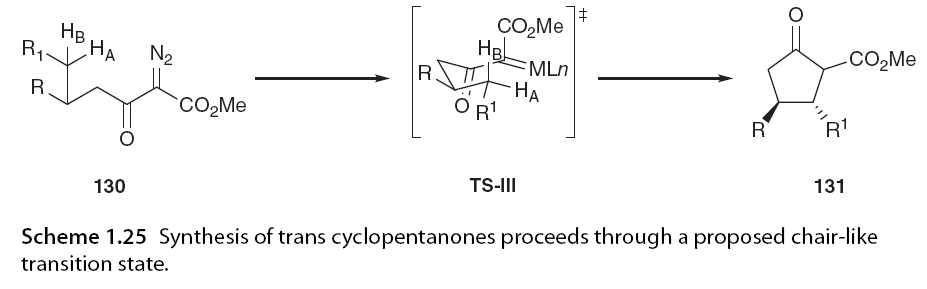
In the C-H insertion reaction of 130, the preference for trans product formation can be explained based on a chair-like transition state (TS-III; Scheme 1.25) where the insertion of C-HA bond yields the trans diastereomeric cyclopentanone 131. The insertion of C-HB bond yields the cis diastereomeric cyclopentanone in a transition state that requires R1 to be in a less favorable axial position [81]. Taber expanded this work by developing a computational model to predict the non-diastereoselectivity of intramolecular C-H insertion reactions based on the cyclization of α-diazo esters (Figure 1.15) [18].
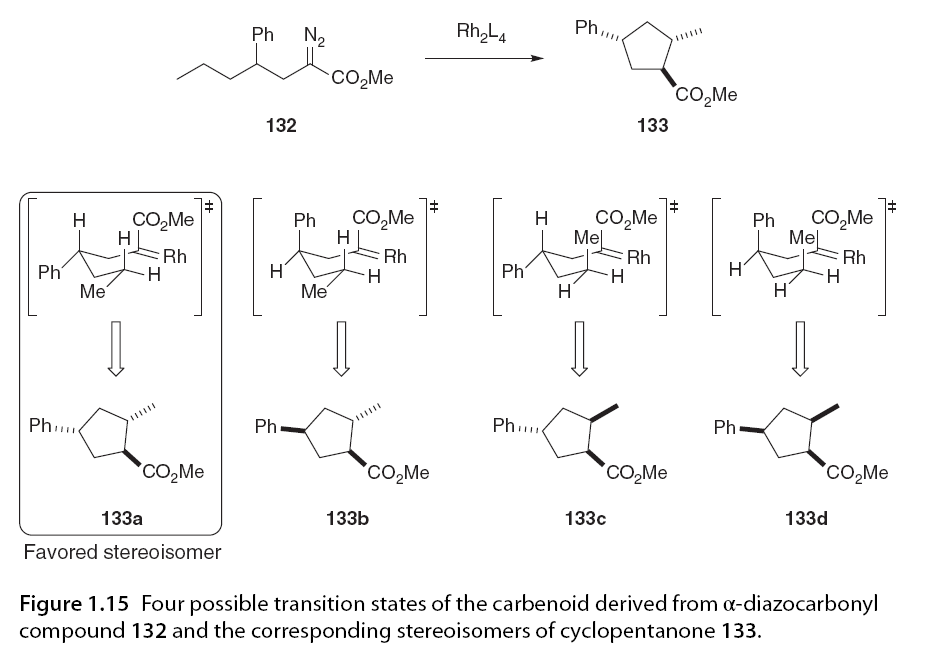
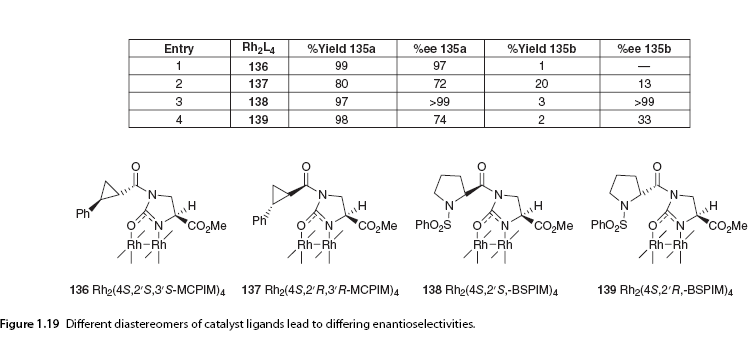
Relative transition state stability analysis of the four possible transition states for the different diastereomers of cyclopentanone 133 was performed. This model was then applied to α-diazo β-keto esters and accurately predicted the formation of the dominant stereoisomer. This study, along with another in-depth theoretical study by Nakamura and colleagues [16], indicates that the trans conformation is preferred in the cyclization reactions of α-diazo esters and α-diazo-β-keto esters due to the alkyl substituents at the insertion site being positioned equatorially in the transition state. In contrast, the cis transition state is subject to 1,3-diaxial repulsion. Nakamura’s calculations emphasized that vinyl or phenyl groups at the insertion site theoretically destabilize the transition state, but only trans products were experimentally observed. 1.5.3.2 Catalyst Effects Doyle later reported excellent cis diastereomeric selectivity and high enantiomeric induction in the chiral rhodium-catalyzed synthesis of bicyclic lactones 135 using Rh2(4S-MACIM)4, while Rh2(5S-MEOX)4 exhibited poorer diastereomeric control in the same system (Scheme 1.26).
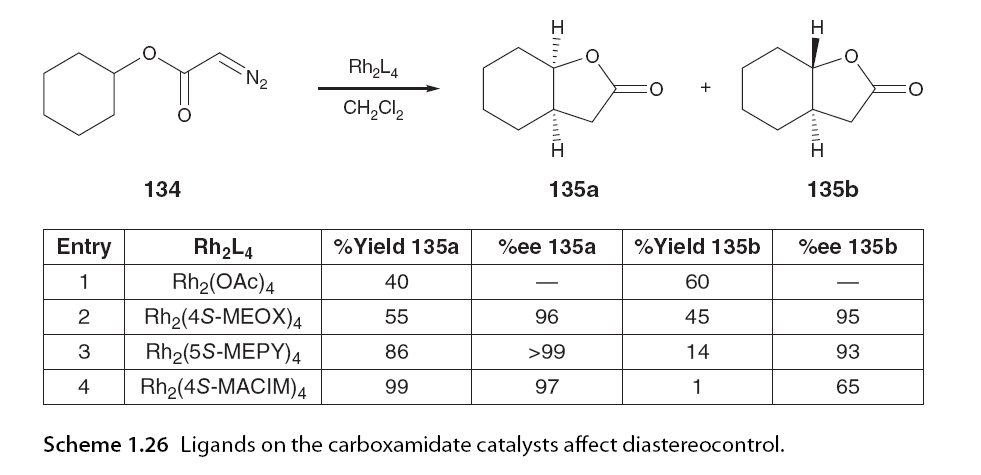
[82]. These examples are just a few among many reactions that highlight the importance of the catalyst on the reaction metal carbene conformation and its resulting stereoselectivity [83-86]. 1.5.4 Understanding Enantiomeric Selectivity The potential for asymmetric induction in intramolecular C-H insertion was first reported by Taber using chiral auxiliaries to preferentially guide the insertion of one face of the carbene. This was followed by the introduction of chiral ligands in dirhodium scaffolds leading to the first reports of catalyzed asymmetric reactions by the McKervey group involving the insertion of C-H in α-diazo carbonyl compounds. To date, numerous studies have reported the influence of various chiral ligands and substrate structures on the enantiomeric selectivity of intramolecular C-H insertion reactions [1,5,6,8]. Symmetry is a major factor in chiral catalysis as it reduces the number of available states in the stereodifferentiation step. Dirhodium paddlewheel structures achieve high symmetry through four identical low molecular weight ligands surrounding a highly symmetric dirhodium core. Taking rhodium with carboxylates as an example, the O-Rh-O plane can be viewed as having an upper and lower surface, with arbitrary α and β (Figure 1.16). Inducing asymmetry in C-H insertion reactions requires the ligands attached to rhodium to restrict the spatial orientation above or below the O–Rh–O plane, allowing only one enantiomeric transition state to be favorable during the reaction, primarily producing one enantiomer. This depends on the orientation of these ligands towards the upper or lower surface of the O-Rh-O plane, allowing the dirhodium complex to adopt various conformations. The symmetry of the complex is also influenced by the inherent symmetry of the ligands. If the ligands are C1 symmetric, asymmetric catalysis is achieved with structure A (resulting in an overall C2 symmetric complex) or B (resulting in an overall D2 symmetric complex) as the planes are equivalent (Figure 1.17) [32].
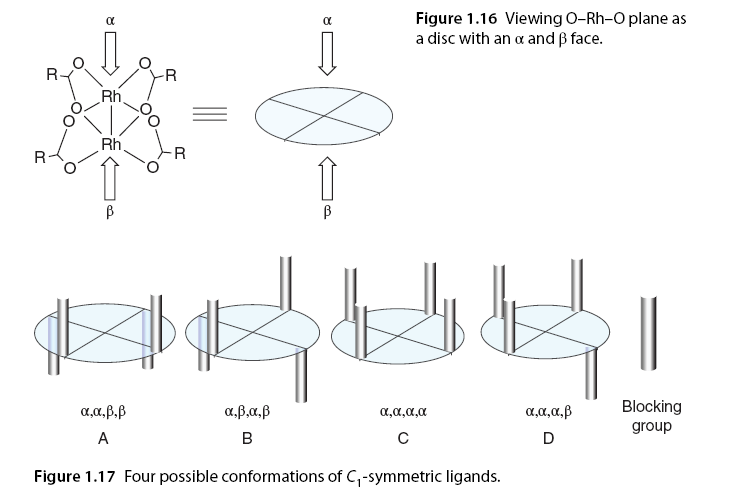
Strong experimental evidence also supports that Rh2(S-PTTL)4 and related catalysts (29-40) participate in asymmetric reactions when the ligands are in conformation C, leading to cyclopropanation, i.e., the “full” or crown conformation, although it was previously thought to be incapable of catalyzing enantioselective reactions [87,88]. While it is challenging to model the flexibility of catalyst ligands experimentally, they are believed to be more flexible than previously thought; this ligand description confirms reports of enhanced enantioselectivity with flexible solvents becoming non-polar solvents such as hexane or pentane [35]. If the carboxylate ligands have C2 symmetry, the catalyst can achieve D4, which is the optimal symmetry for these catalysts, as the two faces of the chiral dirhodium complex are equivalent (conformation E). D2 symmetry can be achieved through two bridging C2 symmetric ligands, such as bridged proline salts, providing a more rigid analogue of C1 symmetric ligands adopting conformation (conformation F) (Figure 1.18).
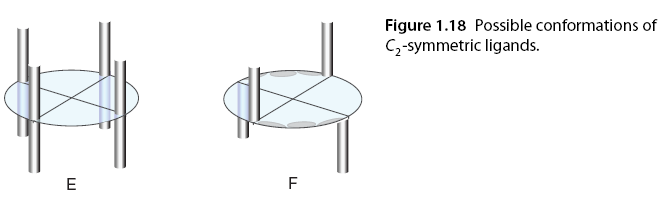
In contrast, the structure of chiral carboxamides is more rigid and is limited to C2 symmetric complex conformations due to their preferred cis (2,2) arrangement. They adopt the α,α,β,β conformation A. Despite the high electronic density, the reactivity of carboxamide ligands is lower than that of carboxylic acid ligands, but they can be more selective in carbene-mediated reactions. To explore the impact of changing the orientation of ligands in space, two sets of diastereomeric catalysts 136/137 and 138/139 were used for the cyclization of α-diazo acetate 134 (Scheme 1.26). Catalysts 136 and 138 produced excellent enantiomeric selectivity, while the use of other isomers of catalysts (137, 139) led to different orientations of ligands resulting in a “matching” conformation or a “mismatched” structure with the substrate [89] (Figure 1.19).
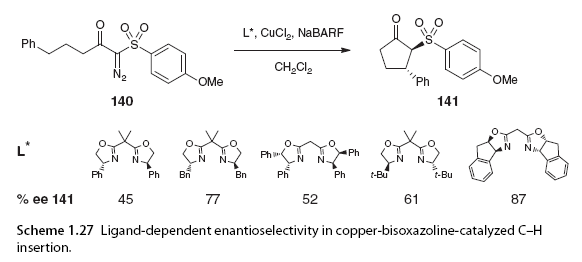
Bisoxazoline ligands have become one of the most widely used ligands due to their C2 symmetric complexes’ ability to catalyze asymmetric reactions with metals such as copper. This C2 symmetry leads to equivalent structures when the catalyst is rotated 180°, meaning that the number of possible transition states and substrate approach trajectories during the reaction is equivalent [90]. While the symmetry of the catalyst is an important factor in producing C-H insertion products with good enantiomeric control, the electronic and spatial properties of the ligands are also crucial. Ligand-dependent enantiomeric selectivity has been reported in studies by the Maguire group involving copper bisoxazoline systems. Substrate 140 is just one example from a recent report on copper-catalyzed C-H insertion of α-diazo carbonyl compounds with five different commercially available bisoxazoline ligands. Significant differences in enantiomeric selectivity were observed among the different ligands used; the best asymmetric induction was achieved using an indole-derived bisoxazoline ligand (87%) (Scheme 1.27) [91].
In addition to the influence of the catalyst on enantiomeric selectivity, the α-diazo carbonyl substrate also plays a significant role. The Maguire group has also demonstrated that variations in the substitution patterns of the two substituents at the insertion site and the α-diazo substituents have a significant impact on enantiomeric control in copper-catalyzed asymmetric C-H insertion. To investigate the effects of substitution at the insertion site, a series of α-diazo carbonyl compounds were prepared in the presence of the catalyst and cyclized using a system composed of bisoxazoline ligands, CuCl2, and the additive sodium 3,5-bis(trifluoromethyl)phenylboronate (NaBARF). The study found that the stereoelectronic characteristics of the insertion site could profoundly affect enantiomeric selectivity depending on the bisoxazoline ligand used. In one example shown here (Scheme 1.28), using a diphenyl bisoxazoline ligand 13, no enantiomeric induction was achieved at the insertion site without a methyl group, while a phenyl group could achieve moderate enantiomeric induction. The enantiomeric selectivity of the benzyl group again decreased.
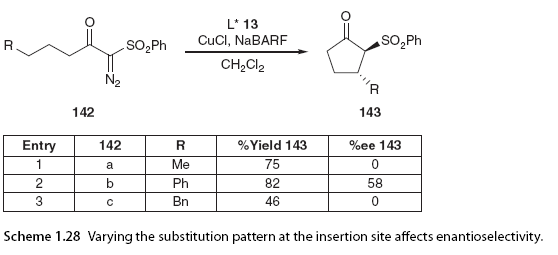
Experiments have shown that the substituents at the insertion site have a significant impact on enantiomeric selectivity; in this case, the reaction with phenyl was found to be optimal [29]. The influence of changing the substitution pattern at the diazo part was also investigated. Initially, various electron-withdrawing groups (sulfones, esters, ketones, ethers, amides, etc.) were studied for their effects on enantiomeric selectivity (Scheme 1.29, entries 1-5).
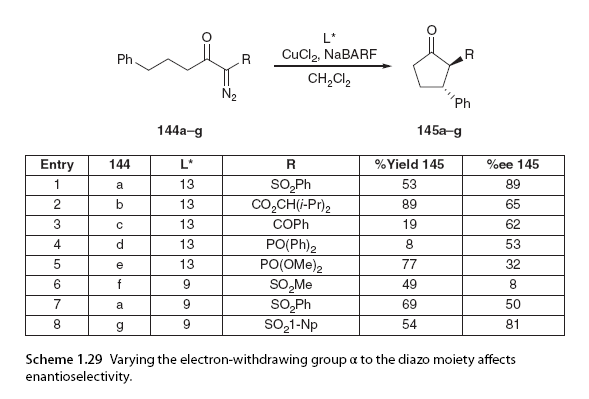
Sulfone functional groups are the best electron-withdrawing groups at the α-diazo site to date. The decline in the electron-withdrawing properties of other groups leads to reduced selectivity and longer reaction times [91,92]. The spatial and electronic effects of the α-diazo motif were also studied with a series of alkyl and aryl sulfonyl substituents. Partial results are shown here (Scheme 1.29, entries 6-8), emphasizing how enantiomeric selectivity changes with increasing steric bulk adjacent to the diazo. This study indicates that electronic effects have a minor impact on asymmetric induction [91]. Furthermore, when using two different materials, different degrees of asymmetric induction were observed with the same substrate (Scheme 1.29, entries 1 and 7). In this example, the diphenyl bisoxazoline ligand in the cyclopentanone product again emphasizes the importance of selecting the optimal catalyst and substrate structure for good enantiomeric control in chemical transformations. Nevertheless, intramolecular C-H insertion has proven to be a key step in the synthesis of various drug-related molecules, some of which are noted here (Figure 1.20) [93-96].
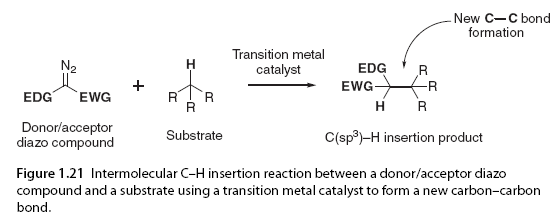
1.6 Intermolecular C(sp3)-H Bond Insertion Intermolecular C-H insertion reactions consist of three components: diazo compounds, substrates that can contain a range of functional groups, and transition metal catalysts (Figure 1.21). This approach facilitates the insertion of heteroatoms into allylic, benzylic, and C(sp3)-H bonds α as simple alkane substrates [97,98]. The selectivity of intermolecular C-H insertion reactions is controlled by steric and electronic factors, with the stabilization of positive charge accumulation during the transition state being key to its synthetic utility (Scheme 1.1 and 1.2). Intermolecular rhodium(II) catalysis using donor/acceptor and acceptor carbene complexes indicates limited selectivity due to the inability to stabilize the highly electrophilic carbene. To achieve the desired selectivity at the required C(sp3)-H sites on intermolecular substrates, donor/acceptor metal carbene is most useful, as the donor group helps stabilize the electron-deficient carbene [99]. To date, most studies have focused on the formation of intermolecular C-C bonds through metal carbene insertion into C(sp3)-H bonds using rhodium(II) catalysts. Therefore, the reactivity and selectivity discussed in this section are primarily based on rhodium(II) catalysts unless otherwise noted.
“In this world, we cannot choose our work, but we can choose our attitude towards it.”
Interpretation: Dickens points out that in real life, many times we may not be able to choose the work we engage in, but we can decide how to approach that work. Whether the work is tedious or challenging, a positive attitude can help us find meaning in our work, improve efficiency, and even derive satisfaction from it; conversely, a negative attitude will only make work more difficult and yield no positive outcomes.
We welcome everyone to like, share, comment, and discuss. Scan the QR code to follow for more exciting content.
