
Academic Exchange Website(www.xueyanhui.com)
Register and member verification can receive up to 1100 yuan coupons
Maximum discount for first order 200 yuan!
Introduction: Dual-atom catalysts (DACs) have tunable electronic structures and spin states, which can enhance the performance of electrochemical reactions. By introducing a second metal, the electronic structure and spin state of the metal center can be altered, allowing for independent regulation of reaction intermediates and improving catalytic activity. Dual-atom catalysts can generally be used for various electrochemical applications, such as CO2 reduction, nitrogen reduction, and oxygen reduction. Through DFT calculations, the electronic structure and spin states of dual-atom catalysts can be studied to understand their catalytic activity and reaction mechanisms. Additionally, these calculations can be used to optimize the structure and performance of dual-atom catalysts, providing theoretical guidance for practical applications. Below, two papers are introduced that illustrate the application of DFT calculations in the study of dual-atom catalysts.
1. Nano Energy: Non-bonding Interactions of Dual-Atom Catalysts for Enhanced Oxygen Reduction Reaction First Author: Mohsen TamtajiCorresponding Authors: Ye Zhu, William A. Goddard III, Wu Wenting, Zhengtang LuoCorresponding Institutions: The Hong Kong Polytechnic University, California Institute of Technology, China University of Petroleum, Hong Kong University of Science and TechnologyThis study demonstrates the design of graphene-supported dual-atom catalysts (DAC) for four-electron oxygen reduction reaction (ORR) using non-bonding interactions of the corresponding metals (M). These non-bonding interactions can synergistically modulate the electronic properties and catalytic activity of the iron active sites in the FeMN6-DAC and FeMN8-DAC systems, where M represents iron, cobalt, nickel, copper, and zinc, respectively. More specifically, when the distance between Fe-M is less than 15 Å, the non-bonding interactions become very significant, making this system a DAC. The predicted ORR overpotentials (ηORR) for FeNiN6-DAC and FeNiN8-DAC are 0.28 V and 0.47 V, respectively, which are at the peak of the volcano plot. This low ηORR is attributed to the high Bader charge transfer and high spin density of the Fe sites in the FeNiN6-DAC and FeNiN8-DAC systems, which weakens the adsorption of the OH* intermediate while enhancing its desorption as H2O. Guided by these density functional theory (DFT) calculation results, FeCoN8-DAC and FeNiN8-DAC were synthesized along with nitrogen-doped graphene. The catalytic activity was verified experimentally, revealing an experimental overpotential of 0.39 V for FeNiN8-DAC with a Tafel slope of 47 mVdec-1. Based on these results, a DFT-guided strategy was proposed to adjust the charge transfer and spin occupancy of active sites, thereby designing DACs for electrochemical ORR.
First Author: Mohsen TamtajiCorresponding Authors: Ye Zhu, William A. Goddard III, Wu Wenting, Zhengtang LuoCorresponding Institutions: The Hong Kong Polytechnic University, California Institute of Technology, China University of Petroleum, Hong Kong University of Science and TechnologyThis study demonstrates the design of graphene-supported dual-atom catalysts (DAC) for four-electron oxygen reduction reaction (ORR) using non-bonding interactions of the corresponding metals (M). These non-bonding interactions can synergistically modulate the electronic properties and catalytic activity of the iron active sites in the FeMN6-DAC and FeMN8-DAC systems, where M represents iron, cobalt, nickel, copper, and zinc, respectively. More specifically, when the distance between Fe-M is less than 15 Å, the non-bonding interactions become very significant, making this system a DAC. The predicted ORR overpotentials (ηORR) for FeNiN6-DAC and FeNiN8-DAC are 0.28 V and 0.47 V, respectively, which are at the peak of the volcano plot. This low ηORR is attributed to the high Bader charge transfer and high spin density of the Fe sites in the FeNiN6-DAC and FeNiN8-DAC systems, which weakens the adsorption of the OH* intermediate while enhancing its desorption as H2O. Guided by these density functional theory (DFT) calculation results, FeCoN8-DAC and FeNiN8-DAC were synthesized along with nitrogen-doped graphene. The catalytic activity was verified experimentally, revealing an experimental overpotential of 0.39 V for FeNiN8-DAC with a Tafel slope of 47 mVdec-1. Based on these results, a DFT-guided strategy was proposed to adjust the charge transfer and spin occupancy of active sites, thereby designing DACs for electrochemical ORR.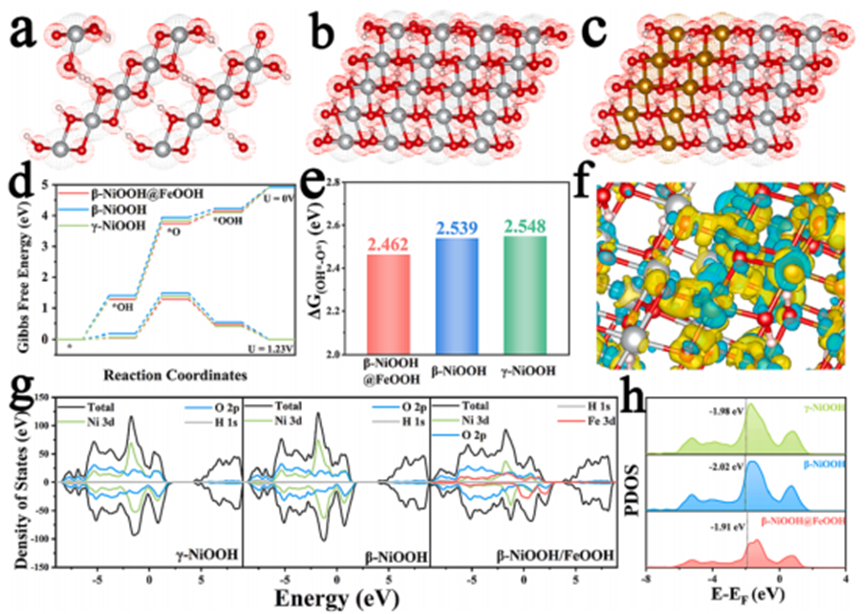 Figure 1. DFT calculation results. (a) Gibbs free energy diagram for the oxygen reduction reaction (ORR) corresponding to the overpotential (ηORR) for FeNiN6-DAC is 0.28 V. The rate-determining step is indicated by the blue circle. The theoretical ηORR for FeMN6-DAC and FeMN8-DAC structures versus the corresponding metals’ d electrons in (b) the volcano plot, indicating that FeNiN6-DAC and FeNiN8-DAC are at the peak. The 3d hybridized orbitals of Fe atoms in FeNiN6-DAC before and after interacting with OH* (c) PDOS, indicating bonding (σ) and antibonding (σ*) orbitals after interaction with OH*. The (d) ηORR of FeMN6-DAC and FeMN8-DAC compared with the new descriptor (φ) indicates that the new descriptor defines a linear relationship between DACs and ηORR.Figure 1a shows the Gibbs free energy diagram for the reaction intermediates of FeNiN6-DAC. Figure 1 shows that the formation of water from OH* is the rate-determining step of the ORR. From Figure 1, it can be seen that the energy barrier and ηORR of FeNiN6-DAC are 0.95 V and |1.23–0.95|=0.28V, respectively. Figure 1b is the volcano plot of the theoretical ηORR, where both FeNiN6-DAC and FeNiN8-DAC are at the peak, with ηORR values of 0.28 V and 0.47 V, respectively, comparable to 0.43V for Pt (111), 0.33V for CoRu@N8V4, and 0.34 for CoCuN6-gra (OH). To further understand the influence of the corresponding metal on the Fe sites’ ORR activity, the projected density of states (PDOS) of the Fe sites was plotted. Figure 1c shows the PDOS of the Fe sites in FeNiN6-DAC before and after interacting with OH*. After interacting with OH*, new peaks are designated as bonding (σ) and antibonding (σ*) orbitals, indicating the interaction between the Fe sites and OH*. According to the results in Figure 1b, the ORR catalytic activity of iron metals strongly depends on the number of d electrons of the corresponding metals. A simple descriptor (φ) is introduced to consider the influence of the most important parameters describing the DACs on the ORR activity:
Figure 1. DFT calculation results. (a) Gibbs free energy diagram for the oxygen reduction reaction (ORR) corresponding to the overpotential (ηORR) for FeNiN6-DAC is 0.28 V. The rate-determining step is indicated by the blue circle. The theoretical ηORR for FeMN6-DAC and FeMN8-DAC structures versus the corresponding metals’ d electrons in (b) the volcano plot, indicating that FeNiN6-DAC and FeNiN8-DAC are at the peak. The 3d hybridized orbitals of Fe atoms in FeNiN6-DAC before and after interacting with OH* (c) PDOS, indicating bonding (σ) and antibonding (σ*) orbitals after interaction with OH*. The (d) ηORR of FeMN6-DAC and FeMN8-DAC compared with the new descriptor (φ) indicates that the new descriptor defines a linear relationship between DACs and ηORR.Figure 1a shows the Gibbs free energy diagram for the reaction intermediates of FeNiN6-DAC. Figure 1 shows that the formation of water from OH* is the rate-determining step of the ORR. From Figure 1, it can be seen that the energy barrier and ηORR of FeNiN6-DAC are 0.95 V and |1.23–0.95|=0.28V, respectively. Figure 1b is the volcano plot of the theoretical ηORR, where both FeNiN6-DAC and FeNiN8-DAC are at the peak, with ηORR values of 0.28 V and 0.47 V, respectively, comparable to 0.43V for Pt (111), 0.33V for CoRu@N8V4, and 0.34 for CoCuN6-gra (OH). To further understand the influence of the corresponding metal on the Fe sites’ ORR activity, the projected density of states (PDOS) of the Fe sites was plotted. Figure 1c shows the PDOS of the Fe sites in FeNiN6-DAC before and after interacting with OH*. After interacting with OH*, new peaks are designated as bonding (σ) and antibonding (σ*) orbitals, indicating the interaction between the Fe sites and OH*. According to the results in Figure 1b, the ORR catalytic activity of iron metals strongly depends on the number of d electrons of the corresponding metals. A simple descriptor (φ) is introduced to consider the influence of the most important parameters describing the DACs on the ORR activity: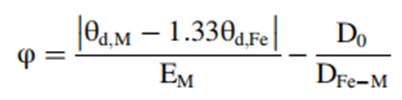 θd is the number of valence electrons in the d orbitals of M, EM is the electronegativity of M, D0 is the nearest distance between Fe and M metals (2.58 A), and DFe-M is the distance between Fe and M metals (from D0 =2.58 A to 15 A). As shown in Figure 1d, the ηORR increases linearly with the descriptor φ.
θd is the number of valence electrons in the d orbitals of M, EM is the electronegativity of M, D0 is the nearest distance between Fe and M metals (2.58 A), and DFe-M is the distance between Fe and M metals (from D0 =2.58 A to 15 A). As shown in Figure 1d, the ηORR increases linearly with the descriptor φ.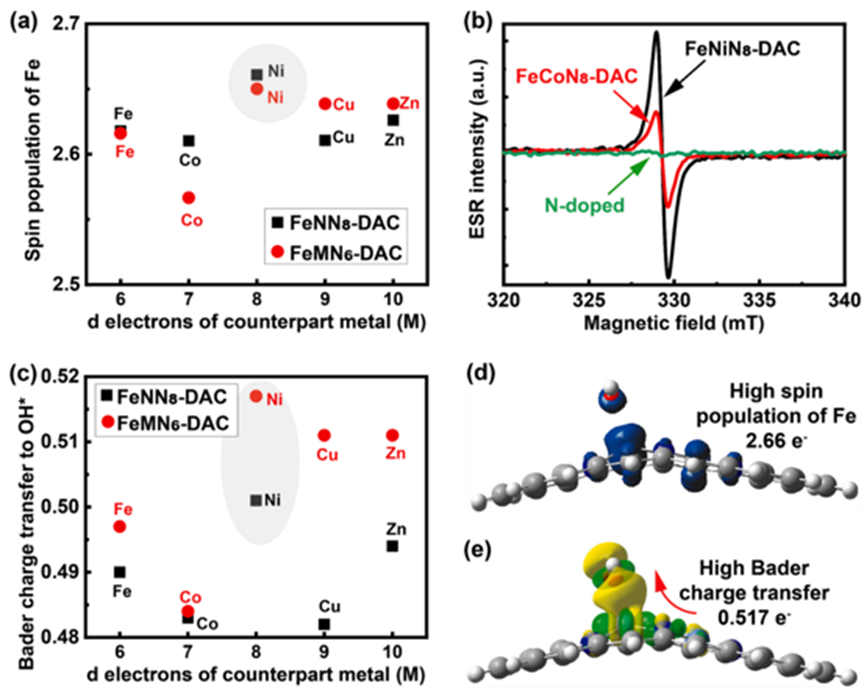 Figure 2 Spin occupancy and Bader charge transfer analysis. (a) Fe atoms (Fe-OH*) with corresponding metals (M) spin density, indicating that FeNiN6-DAC and FeNiN8-DAC have high spin densities. The electronic spin resonance (ESR) spectrum of the samples measured at 77 K shows that the spin density of FeNiN8-DAC is higher than that of FeCoN8-DAC, while the spin density of nitrogen-doped samples shows no peak. (c) Bader charge transfer from the Fe sites to the OH* intermediate, relative to the corresponding metals’ d electrons. (d) Spatial spin density of FeNiN6-DAC (isovalue = 0.015 e/A3). Blue and green represent alpha and beta spin densities, respectively. (e) Side view of charge transfer within the FeNiN6-DAC system (isovalue = 0.004 e/A3). Yellow and green represent charge accumulation and dispersion, respectively. This indicates that the FeNiN6-DAC system has high spin occupancy and high charge transfer capability. Relatively high charge transfer from Fe to Ni increases the oxidation state of Fe, weakening the adsorption of the *OH intermediate, and reducing the overpotential of ORR by increasing the spin density of the Fe sites (Figure 2a). The results are consistent with the electronic spin resonance (ESR) spectral results of nitrogen-doped, FeCoN8-DAC, and FeNiN8-DAC at 77 K, as shown in Figure 2b. Figure 2c shows the high charge transfer between FeNiN6-DAC and the OH* intermediate. Figures 2d-e show the high spin polarization of the Fe sites in FeNiN6-DAC, but its high charge transfer promotes the desorption of OH* to release water, making this structure a promising ORR catalyst.References:Tamtaji M, Peng Q, Liu T, et al. Non-bonding interaction of dual atom catalysts for enhanced oxygen reduction reaction[J]. Nano Energy, 2023, 108: 108218.
Figure 2 Spin occupancy and Bader charge transfer analysis. (a) Fe atoms (Fe-OH*) with corresponding metals (M) spin density, indicating that FeNiN6-DAC and FeNiN8-DAC have high spin densities. The electronic spin resonance (ESR) spectrum of the samples measured at 77 K shows that the spin density of FeNiN8-DAC is higher than that of FeCoN8-DAC, while the spin density of nitrogen-doped samples shows no peak. (c) Bader charge transfer from the Fe sites to the OH* intermediate, relative to the corresponding metals’ d electrons. (d) Spatial spin density of FeNiN6-DAC (isovalue = 0.015 e/A3). Blue and green represent alpha and beta spin densities, respectively. (e) Side view of charge transfer within the FeNiN6-DAC system (isovalue = 0.004 e/A3). Yellow and green represent charge accumulation and dispersion, respectively. This indicates that the FeNiN6-DAC system has high spin occupancy and high charge transfer capability. Relatively high charge transfer from Fe to Ni increases the oxidation state of Fe, weakening the adsorption of the *OH intermediate, and reducing the overpotential of ORR by increasing the spin density of the Fe sites (Figure 2a). The results are consistent with the electronic spin resonance (ESR) spectral results of nitrogen-doped, FeCoN8-DAC, and FeNiN8-DAC at 77 K, as shown in Figure 2b. Figure 2c shows the high charge transfer between FeNiN6-DAC and the OH* intermediate. Figures 2d-e show the high spin polarization of the Fe sites in FeNiN6-DAC, but its high charge transfer promotes the desorption of OH* to release water, making this structure a promising ORR catalyst.References:Tamtaji M, Peng Q, Liu T, et al. Non-bonding interaction of dual atom catalysts for enhanced oxygen reduction reaction[J]. Nano Energy, 2023, 108: 108218.
DOI: 10.1016/j.nanoen.2023.108218
2. Chem. Eng. J: Heteronuclear Ni-Ag Dual-Atom Catalysts for Efficient CO2 Electroreduction with Nearly 100% CO Selectivity
First Author: Zeyu Guo
Corresponding Authors: Wu Tao, Xu Mengxia
Corresponding Institution: University of Nottingham Ningbo China
Single-atom catalysts (SAC) have become highly attractive materials in the field of electrochemical carbon dioxide reduction (ECO2R). Dual-atom catalysts (DAC) are an extension of single-atom catalysts, exhibiting more remarkable functions due to the synergistic effects between adjacent metal atoms. However, reasonable design, clear coordination patterns, and a deep understanding of the synergistic mechanisms of heteronuclear dual-atom catalysts remain unknown. This paper synthesizes a heteronuclear Ni-Ag dual-atom catalyst supported on nitrogen-rich porous carbon using a cascade pyrolysis method, named Ni-Ag/PC-N. The configuration of the Ni-Ag dual-atom sites was confirmed to be N3-Ni-Ag-N3. The Ni-Ag/PC-N exhibits over 90% Faradaic efficiency (FECO) for CO2 reduction over a wide range of applied potentials (i.e., from -0.7 to -1.3 V relative to the reversible hydrogen electrode (RHE)). At -0.8 V (relative to the reversible hydrogen electrode), the peak FECO reaches 99.2%. Tafel analysis indicates that the rate-determining step for the conversion of ECO2R to CO is the formation of the *COOH intermediate, and Ni-Ag/PC-N exhibits the best electrokinetic properties. In situ Fourier-transform infrared spectroscopy and in situ Raman spectroscopy indicate that the generation rate of the *COOH intermediate accelerates during the conversion of ECO2R to CO. Density functional theory (DFT) calculations show that the coordinated nickel atoms lower the energy barrier for the formation of the *COOH intermediate on the Ni-Ag/PC-N surface, while the adjacent silver atoms alleviate the strong *CO affinity on the nickel sites, preventing catalyst poisoning.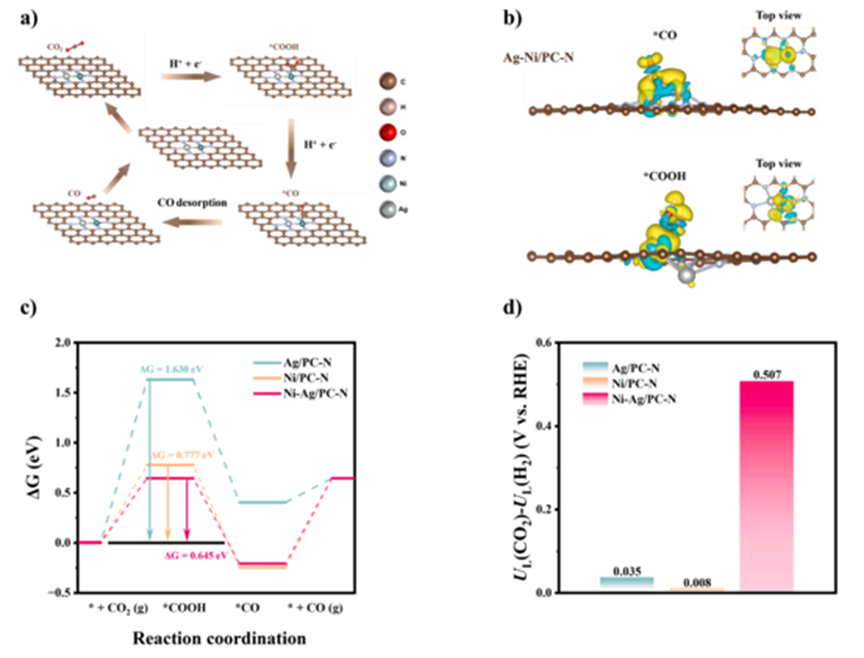
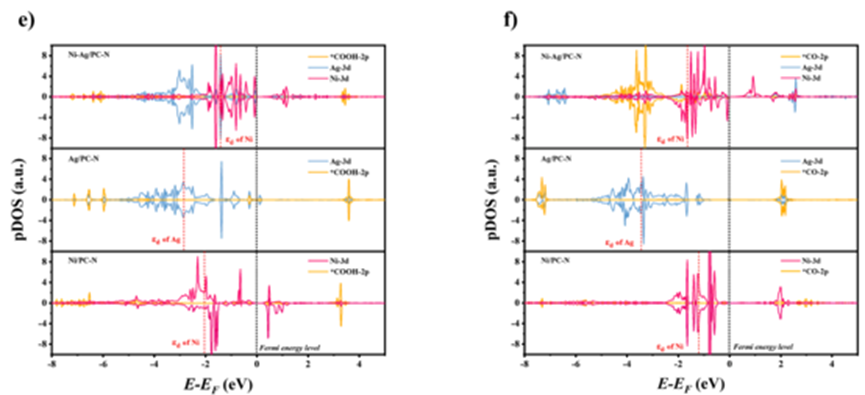 Figure 3 (a) Schematic diagram of the mechanism for the conversion of ECO2R to CO on N3-Ni*-Ag-N3; (b) Charge density differences of the intermediate products *CO and *COOH on the Ni-Ag/PC-N, Ag/PC-N, and Ni/PC-N sites, with an isovalue set to 0.002 e/Å3, yellow and cyan representing electron accumulation and depletion, respectively; (c) Gibbs free energy diagram for the process of ECO2R to CO; (d) Theoretical limiting potential differences for ECO2R and HER on Ag/PC-N, Ni/PC-N, and Ni-Ag/PC-N; (e&f) Projected density of states of Ni/Ag-3d and C-2p orbitals for different adsorptions of *CO/*COOH intermediates. To reveal the fundamental reasons for the enhanced activity of adjacent Ni-Ag sites in ECO2R, DFT calculations were performed. Specifically, based on the coordination of N3-Ni-Ag-N3, the adsorption energies of the intermediates on the Ni (N3-Ni*-Ag-N3), Ag (N3-NiAg*-N3), and Ni-Ag (N3-Ni*-Ag*-N3) coordination patterns were simulated. As shown in Figure 3a, the optimal mode with the lowest free energy was identified as N3-Ni*-Ag-N3. Furthermore, the charge density differences of key intermediates (*COOH and *CO) were calculated, as shown in Figure 3b. This demonstrates that the dual-atom Ni-Ag pair exhibits electronic interactions with the coordinated N atoms and the *CO/*COOH intermediates. This observation strongly indicates that the addition of the Ni-Ag dual-atom pair enhances the stability of the adsorption of *CO/*COOH and effectively reduces the energy barriers associated with the entire process. According to Figure 3c, during the step of converting ECO2R to CO, the formation of the *COOH species is an endothermic process, with energy barriers of N3-Ni*-Ag-N3, Ni-N3, and Ag-N3 being 0.64 eV, 0.78 eV, and 1.63 eV, respectively, indicating that the formation of *COOH is the ratedetermining step. Compared to Ni-N3 and Ag-N3, the heteronuclear Ni-Ag/PCN dual-atom sites reduce the energy barriers by 0.14 eV and 0.99 eV, respectively, thereby enhancing the catalytic activity.HER and ECO2R electronic selectivity can be quantified using the UL(CO2)-UL(H2) values, where UL(CO2) and UL(H2) denote the theoretical limiting potentials for ECO2R and HER, respectively (Figure 6d). The Ni-Ag/PC-N has the highest UL(CO2)-UL(H2) value (0.507 V), followed by Ag/PC-N (0.035 V) and Ni/PC-N (0.008 V). These computational results indicate that the Ni-Ag/PC-N exhibits the best suppression effect on HER and the highest selectivity for ECO2R, which is consistent with experimental observations such as FEs, ECSA, and OH- adsorption tests. To understand the interactions between the reaction intermediates and the sites, the projected density of states of the Ni-Ag/PC-N, Ni/PC-N, and Ag/PC-N for the Ni-3d, Ag-3d, and C-2p orbitals were calculated (pDOS). The results shown in Figures 3e and f indicate significant hybridization between the Ni-3d, Ag-3d, and C-2p orbitals, suggesting strong bonding interactions in the coordination environment. Furthermore, the d-band center of Ni in the adsorbed *COOH on the heteroatom Ni-Ag/PC-N (-1.41 eV) is closer to the Fermi level. This may be due to charge redistribution caused by the introduction of Ag, leading to a narrowing of the d-band gap of the Ni species, thus improving electron mobility and accelerating the formation of *COOH. However, the d-band center of Ni in the adsorbed *CO on the Ni-Ag/PC-N shifts from down to -1.59 eV, indicating a weakened electron transfer capability. This suggests that the binding affinity of the *CO intermediate at the N3-Ni*-Ag-N3 site is reduced, decreasing the resistance to desorption of the CO molecule, which aligns well with the charge density difference and free energy calculation results.References:Guo, Zeyu, et al. “Synergistic engineering of heteronuclear Ni-Ag dual-atom catalysts for high-efficiency CO2 electroreduction with nearly 100% CO selectivity.” Chemical Engineering Journal 476 (2023): 146556.
Figure 3 (a) Schematic diagram of the mechanism for the conversion of ECO2R to CO on N3-Ni*-Ag-N3; (b) Charge density differences of the intermediate products *CO and *COOH on the Ni-Ag/PC-N, Ag/PC-N, and Ni/PC-N sites, with an isovalue set to 0.002 e/Å3, yellow and cyan representing electron accumulation and depletion, respectively; (c) Gibbs free energy diagram for the process of ECO2R to CO; (d) Theoretical limiting potential differences for ECO2R and HER on Ag/PC-N, Ni/PC-N, and Ni-Ag/PC-N; (e&f) Projected density of states of Ni/Ag-3d and C-2p orbitals for different adsorptions of *CO/*COOH intermediates. To reveal the fundamental reasons for the enhanced activity of adjacent Ni-Ag sites in ECO2R, DFT calculations were performed. Specifically, based on the coordination of N3-Ni-Ag-N3, the adsorption energies of the intermediates on the Ni (N3-Ni*-Ag-N3), Ag (N3-NiAg*-N3), and Ni-Ag (N3-Ni*-Ag*-N3) coordination patterns were simulated. As shown in Figure 3a, the optimal mode with the lowest free energy was identified as N3-Ni*-Ag-N3. Furthermore, the charge density differences of key intermediates (*COOH and *CO) were calculated, as shown in Figure 3b. This demonstrates that the dual-atom Ni-Ag pair exhibits electronic interactions with the coordinated N atoms and the *CO/*COOH intermediates. This observation strongly indicates that the addition of the Ni-Ag dual-atom pair enhances the stability of the adsorption of *CO/*COOH and effectively reduces the energy barriers associated with the entire process. According to Figure 3c, during the step of converting ECO2R to CO, the formation of the *COOH species is an endothermic process, with energy barriers of N3-Ni*-Ag-N3, Ni-N3, and Ag-N3 being 0.64 eV, 0.78 eV, and 1.63 eV, respectively, indicating that the formation of *COOH is the ratedetermining step. Compared to Ni-N3 and Ag-N3, the heteronuclear Ni-Ag/PCN dual-atom sites reduce the energy barriers by 0.14 eV and 0.99 eV, respectively, thereby enhancing the catalytic activity.HER and ECO2R electronic selectivity can be quantified using the UL(CO2)-UL(H2) values, where UL(CO2) and UL(H2) denote the theoretical limiting potentials for ECO2R and HER, respectively (Figure 6d). The Ni-Ag/PC-N has the highest UL(CO2)-UL(H2) value (0.507 V), followed by Ag/PC-N (0.035 V) and Ni/PC-N (0.008 V). These computational results indicate that the Ni-Ag/PC-N exhibits the best suppression effect on HER and the highest selectivity for ECO2R, which is consistent with experimental observations such as FEs, ECSA, and OH- adsorption tests. To understand the interactions between the reaction intermediates and the sites, the projected density of states of the Ni-Ag/PC-N, Ni/PC-N, and Ag/PC-N for the Ni-3d, Ag-3d, and C-2p orbitals were calculated (pDOS). The results shown in Figures 3e and f indicate significant hybridization between the Ni-3d, Ag-3d, and C-2p orbitals, suggesting strong bonding interactions in the coordination environment. Furthermore, the d-band center of Ni in the adsorbed *COOH on the heteroatom Ni-Ag/PC-N (-1.41 eV) is closer to the Fermi level. This may be due to charge redistribution caused by the introduction of Ag, leading to a narrowing of the d-band gap of the Ni species, thus improving electron mobility and accelerating the formation of *COOH. However, the d-band center of Ni in the adsorbed *CO on the Ni-Ag/PC-N shifts from down to -1.59 eV, indicating a weakened electron transfer capability. This suggests that the binding affinity of the *CO intermediate at the N3-Ni*-Ag-N3 site is reduced, decreasing the resistance to desorption of the CO molecule, which aligns well with the charge density difference and free energy calculation results.References:Guo, Zeyu, et al. “Synergistic engineering of heteronuclear Ni-Ag dual-atom catalysts for high-efficiency CO2 electroreduction with nearly 100% CO selectivity.” Chemical Engineering Journal 476 (2023): 146556.
DOI: 10.1016/j.cej.2023.146556.
3. Angew. Chem. Int. Ed. Cu-Co Dual-Atom Catalysts Triggered Water Activation for Silane Oxidation

First Author: Liping ZhangCorresponding Authors: Zhao Peiqing, Cui XinjiangCorresponding Institution: Lanzhou Institute of Chemical Physics, Chinese Academy of SciencesA high-performance catalyst that can significantly lower the water activation energy barrier is crucial for promoting reactions limited by water dissociation. This study proposes a Cu-Co dual-atom catalyst (CuCo-DAC) with a uniform and clear CuCoN6(OH) structure that jointly promotes water activation during silane oxidation. The catalytic performance of this catalyst far exceeds that of single-atom catalysts (SAC). The yield of various functional silanes converted to silanol reaches up to 98%, with a selectivity of up to 99%. Kinetic studies indicate that the activation energy for silane oxidation of the CuCo-DAC is significantly lower than that of the Cu single-atom catalyst (Cu-SAC) and Co single-atom catalyst (Co-SAC). Theoretical calculations prove two different reaction pathways, where water splitting is the rate-determining step, and the CuCo-DAC accelerates this process, while the formation of H2 is the key step for single-atom catalysts.
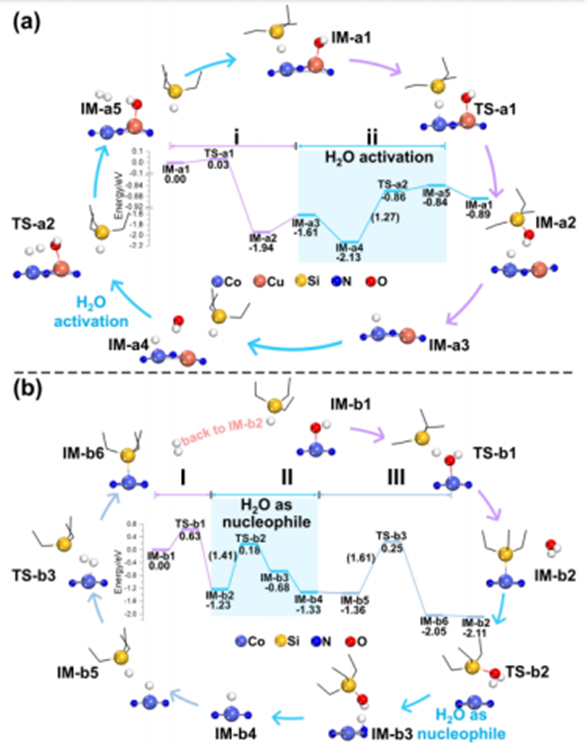
Figure 1 (a) Simulation paths and potential energy surfaces for silane oxidation on CuCo-DAC and (b) Co-SAC. Note: TS and IM represent transition states and intermediates, respectively. Using triethylsilane (Et3SiH) as a model molecule, the reaction mechanism was studied through DFT calculations. Figure 1a shows the optimal potential energy surface of CuCo-DAC. The reaction path can be divided into two steps: OH removal (step i: IM-a1 to IM-a3) and H2O activation (step ii: IM-a3 to IM-a5) (Figure 1a). In step i, Et3SiH is initially adsorbed at the Co site, with OH on Cu, and then OH directly attacks the Si atom, breaking the Si-H bond and forming Si-OH bond, with an energy barrier as low as 0.03eV (IM-a1 to TS-a1), and a strong exothermic reaction of 1.94eV (IM-a1 to IM-a2, reaction a1). Meanwhile, the CoH intermediate is transformed during the formation of the silanol, with an energy lower than that of the formation of CuH. After silanol desorption, step ii begins with the adsorption of H2O, which reacts with CoH to produce CoH2 and CuOH, with energy barriers and reaction energies of 1.27 (IM-a4 to TS-a2) and 1.29eV (IM-a4 to IM-a5) (reaction a2). Notably, in the presence of Cu-SAC and Co-SAC, completely different reaction mechanisms were observed for silane oxidation. As shown in Figure 4b, this includes three steps: OH removal (step I: IM-b1 to IM-b2), H2O acting as a nucleophile (step II: IM-b2 to IM-b4), and H2 generation (step III: IM-b4 to IM-b6). First, the hydroxyl group on CoOH is removed through reaction with Et3SiH, generating Et3Si Co and H2O, with energy barrier and reaction energy of 0.63eV (IM-b1 to TS-b1) and 1.23eV (IM-b1 to IM-b2) (reaction b1). Then in the second step, H2O acts as a nucleophile directly attacking the Et3SiCo bond, forming the product Et3SiOH and H species, with an energy barrier of 1.41eV (IM-b2 to TS-b2), with an endothermic process of 0.65eV (IM-b3 to IM-b4, reaction b2). It should be noted that in step III, the H species is more likely to be stabilized by adjacent surface atoms (denoted as NsH). Therefore, in the case of CuCo-DAC, the deactivation of H2 (step ii) is the rate-determining step, whereas in Co-SAC and Cu-SAC, the reaction is limited by H2 generation (step III). As the barriers increase sequentially to CuCo-DAC (1.27eV), Co-SAC (1.61eV), and Cu-SAC (2.03eV), the catalytic performance of Cu-CoDAC is the highest, which is consistent with experimental results. More importantly, Cu-SAC and Co-SAC directly use H2O as a nucleophile, while CuCo-DAC effectively dissociates it, providing a theoretical basis for the difference in catalytic activity.ReferencesZhang, Li, et al. “Water Activation Triggered by Cu− Co Double‐Atom Catalyst for Silane Oxidation.” Angewandte Chemie 135.47 (2023): e202313343.
DOI: 10.1002/anie.202313343
4. APPL CATAL B-ENVIRON: Controlled Synthesis of Ni2 Dual-Atom Catalysts for Synergistic CO2 Electroreduction

First Author: Xiang-Ming LiangCorresponding Author: Lu Tongbu
Corresponding Institution: Tianjin University of Technology
Dual-atom catalysts (DAC) have attracted much attention due to the synergistic effects within their dual-atom sites, but the controlled synthesis of pure dual-atom catalysts remains a challenge. This paper uses a bimetallic nickel complex as a precursor to uniformly anchor directly bonded nickel pairs on nitrogen-doped carbon nanotubes, achieving the controlled synthesis of uniform dual-atom nickel catalysts (Ni2-NCNT). The Ni2-NCNT exhibits extraordinary catalytic activity, selectivity, and stability in the electroreduction of CO2 to CO, with a partial current density (jCO) of 76 mA cm-2. The mass activity of Ni2-NCNT is 2.3 times that of the corresponding single-atom catalyst (Ni1-NCNT). DFT calculations indicate that the formation of the *COOH intermediate is significantly facilitated by the synergistic coordination at the Ni2 dual-atom sites, thereby promoting the electroreduction of CO2.
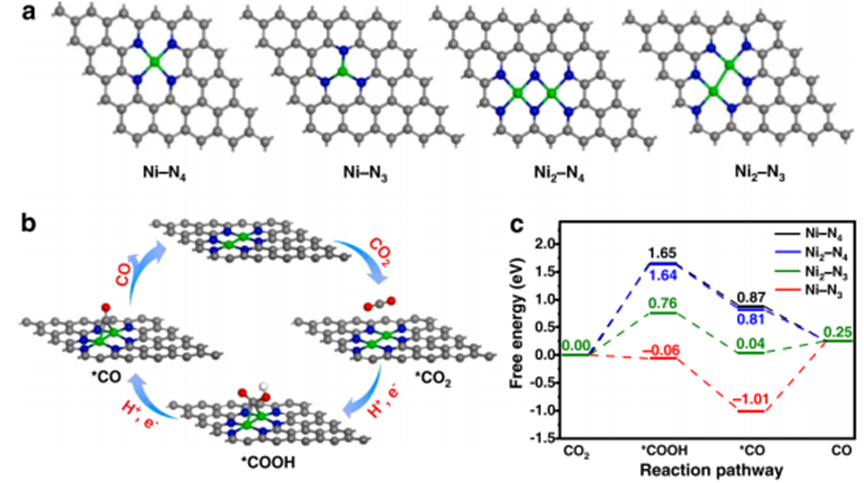
Figure 2 (a) Structural models of Ni-N4 (Ni1-NCNT) and Ni-N3 SAC as well as Ni2-N4 and Ni2-N3 (Ni2-NCNT) DAC. (b) Proposed reaction pathway for the electroreduction of CO2 to CO by Ni2-N3 (Ni2-NCNT). (c) Calculated Gibbs free energy diagram for the conversion of CO2 to CO. To further reveal the mechanism of CO2RR and the activity differences between Ni2-NCNT and Ni1-NCNT, density functional theory (DFT) calculations were performed. For comparison, two optimized DAC models of Ni2-N4 and Ni2-N3 (Ni2-NCNT) were constructed, as well as two corresponding SAC models of Ni-N4 (Ni1-NCNT) and Ni-N3 (Figure 2a). As shown in Figure 2b, the process of electroreduction of CO2 to CO involves four fundamental steps: (1) CO2 + * → *CO2 ( * = catalytic site); (2) *CO2 + H+ + e- → *COOH; (3) *COOH + H+ + e- → *CO + H2O; (4) *CO → CO + *. The Gibbs free energy diagrams for these four steps are shown in Figure 2c. In the steps of CO2 adsorption and proton-coupled electron transfer (PCET), the free energy changes (ΔG) for Ni-N4, Ni2-N4, Ni2-N3, and Ni-N3 are 1.65 eV, 1.64 eV, 0.76 eV, and -0.06 eV, respectively, with Ni-N3 showing the most favorable ΔG for forming the adsorbed *COOH intermediate. Subsequently, the *COOH intermediate undergoes a second PCET, forming water molecules and *CO, with a downward trend in free energy change for all four catalyst models. Finally, *CO desorbs from the catalyst and releases the active site. For Ni-N3 and Ni2-N3, the *CO desorption step is endothermic, with free energy changes of 1.26 eV and 0.21 eV, respectively. For Ni-N4 and Ni2-N4, the *CO desorption step is spontaneous, with free energy changes of -0.62 eV and -0.56 eV, respectively. According to the calculations, the formation of the *COOH intermediate in Ni-N4, Ni2-N4, and Ni2-N3 is regarded as the rate-determining step (RDS), while the desorption of the *CO intermediate is the RDS for Ni-N3. Among the four catalyst models, Ni2-N3 exhibits the highest electrochemical activity for carbon dioxide reduction, with a free energy change of 0.76 eV for the rate-determining step, and the synergistic effect between the two Ni atoms in Ni2-N3 stabilizes the *COOH intermediate, significantly reducing the free energy change of the RDS compared to Ni-N4 SAC (1.65 eV). Therefore, Ni2-N3 exhibits the best electrochemical activity for carbon dioxide reduction, which is consistent with experimental results.ReferencesLiang, Xiang-Ming, et al. “Controlled synthesis of a Ni2 dual-atom catalyst for synergistic CO2 electroreduction.” Applied Catalysis B: Environmental 322 (2023): 122073.
DOI: 10.1016/j.apcatb.2022.122073
Welcome to join the Academic Exchange QQ Group: 701960961. The purpose of this group is to provide a platform for gathering researchers, software developers, and enthusiasts in quantum chemistry, first-principles calculations, molecular dynamics, machine learning, and other fields to promote and disseminate theoretical calculations. Everyone is welcome to invite more people interested in theoretical calculations to join the group for communication and learning!
Theoretical Calculations | Simulation and Modeling
One-on-one Contact

Teacher Pang
188 1363 0548
www.xueyanhui.com