
Previously, we have laid some groundwork for your understanding of microbiomes (see the review at the end of the article), and we will also update you with tutorials on amplicon sequencing, metagenomics, and single-cell microbiome analysis, so stay tuned!

1. Introduction to QIIME2
QIIME2 is a powerful, extensible, and decentralized microbiome analysis platform that integrates various analytical functions. Since its launch in 2019 in Nat Biotechnol, it has quickly gained popularity, with total visits exceeding 53,000 and total citations reaching over 7,000. This article will kick off a practical analysis of 16S rDNA sequencing data based on this platform. For an introduction to the functions and processes of the QIIME2 platform, please refer to Professor Liu Yongxin’s blog.


2. Installing QIIME2
# Ensure you are running in a Linux or other Unix-like environment
# Update miniconda (optional)
conda update conda
# Install wget (optional)
conda install wget
# Download the required software list and build a conda environment named `qiime2-2022.2`, the time required depends on network speed and can be resumed.
## Tip1: The software list `qiime2-2022.2-py38-linux-conda.yml` can also be accessed and downloaded via the browser address bar, note the download path, and the file extension must be `.yml`.
wget https://data.qiime2.org/distro/core/qiime2-2022.2-py38-linux-conda.yml
# View the software list and count the number of lines (optional)
head -n 10 qiime2-2022.2-py38-linux-conda.yml
## channels:
## - qiime2/label/r2022.2
## - https://packages.qiime2.org/qiime2/2022.2/passed/core/
## - conda-forge
## - bioconda
## - defaults
## dependencies:
## - _libgcc_mutex=0.1
## - _openmp_mutex=4.5
## - _r-mutex=1.0.1
# Count the number of lines in the software list to estimate the number of dependencies (the number of times "-" appears minus the number of channels)
grep -v "-" qiime2-2022.2-py-linux-conda.yml | wc -l
## 2, meaning there are only two lines without "-", which are "channels:" and "dependencies:"
wc -l qiime2-2022.2-py-linux-conda.yml
## 460, meaning the qiime2-2022.2-py-linux-conda.yml file has a total of 460 lines,
## that is, excluding channels, there are 453 dependent software...(⊙o⊙)By default, Miniconda/Anaconda will be installed in the home directory (home/ or ~/), and the conda-built environment <span>qiime2-2022.2</span> will appear in the <span>Miniconda</span> directory under the <span>envs</span> directory.
#Tip2: The official recommendation is to install via conda, considering the large number of software used, using conda will take a long time (at least starting from 90 min/(ㄒoㄒ)/~~), here we replace ‘conda’ with ‘mamba’ to speed up the download. Also, run it in the background. Here we first install mamba as recommended by the official documentation, and then use mamba for QIIME2 environment configuration.It was tested that using mamba took about 42 min 42 s.
# Install mamba to the base environment for easier subsequent use.
conda install mamba -n base -c conda-forge
time mamba env create -n qiime2-2022.2 --file qiime2-2022.2-py38-linux-conda.yml &
## real 42m41.873s
## user 6m51.199s
## sys 2m45.458sHere we used conda to install mamba as a program call, taking about 43 min in total. This shows one of the most common usages of mamba, which is itself a package manager similar to conda; for more detailed usage, please refer to the mamba documentation.<span>Micromamba</span> is a minimal version of the Mamba package manager. It is a pure C++ package with a separate command-line interface. It can be used as an alternative to <span>Miniconda</span> for bootstrapping environments. Since the above installation took about 43 min, which is still a bit long, we will try to use <span>micromamba</span> instead of <span>conda</span> for environment configuration.
# Download and extract micromamba
wget -qO- https://micro.mamba.pm/api/micromamba/linux-64/latest | tar -xvj bin/micromamba
# At this point, directly calling micromamba will show the following prompt:
micromamba activate---'micromamba' is running as a subprocess and can't modify the parent shell. Thus you must initialize your shell before using activate and deactivate. To initialize the current bash shell, run: $ eval "$(micromamba shell hook --shell=bash)" and then activate or deactivate with: $ micromamba activate
To automatically initialize all future (bash) shells, run: $ micromamba shell init --shell=bash --prefix=~/micromamba
Supported shells are {bash, zsh, xonsh, cmd.exe, powershell, fish}.
critical libmamba Shell not initialized*---# The reason is mainly that I am in the conda environment, and micromamba as a subprocess cannot change the configuration of the parent process conda.
# So according to the prompt, temporarily activate micromamba in this terminal and set it using 'eval'.
eval "$(micromamba shell hook --shell=bash)"
# Of course, you can also set it to activate automatically, but this way, the automatically called one in subsequent terminals will be micromamba instead of conda.
micromamba shell init --shell=bash --prefix=~/micromamba
micromamba activate # Here we will name the new environment qiime2-2022.2 and use micromamba to download and install. The actual time taken was about 23 min 27 s, and the time was shortened a bit :)time micromamba create -n qiime2-2022.2 -f qiime2-2022.2-py38-linux-conda.yml &
## real 23m27.549s
## user 8m39.211s
## sys 1m43.348s
# Activate the qiime2 environment
micromamba activate qiime2-2022.2
qiime --help
3. Selecting Data
For this data analysis, we will select a paper published in 2021 in Cell Mol Gastroenterol Hepatol regarding the role of the aryl hydrocarbon receptor in intestinal epithelial cells in influencing gut microbiota and intervening in the development of alcohol-related liver disease in mice. The authors constructed AhrΔIEC and Ahrfl/fl mice and intervened with alcohol or control feed to observe changes in the gut microbiota of the mice. A total of 28 samples of cecal contents from mice were used in this study, which has a small data volume, making it easy to download and analyze, suitable for beginners.


4. Downloading Sequences from SRA Archive
The 16S rDNA sequences from this paper have been uploaded to the NCBI SRA (Sequence Read Archive) database, with the accession number PRJNA663684, which can be downloaded publicly. The SRA database is used to store raw data from second-generation sequencing, and the data included can be divided into four categories:
-
Studies — Research topics: objectives and experimental goals, often prefixed with ERP, SRP, etc., and more recently with PRJNA.
-
Experiments — Experimental designs: containing information such as Sample, DNA source, sequencing platform, data processing, etc., often prefixed with SRX.
-
Runs — Sequencing result sets: representing the reads generated by the sequencer, prefixed with SRR.
-
Samples — Sample information: including collection time, other information about patients or animals, etc.
The usual practice for downloading data from SRA is to use the <span>prefetch</span> function provided by the sra-tools component from NCBI to obtain the <span>.sra</span> files, and then use <span>fasterq-dump</span> or <span>fastq-dump</span> for conversion to obtain sequence files. Below is an example process of downloading using this workflow:
First, prepare the required software and data list —
# Install sra-tools component
micromamba install sra-tools
mkdir -p ./qiime2_test/ALD_mice; cd ./qiime2_test/ALD_mice
# Prepare data list
## Use SRA run selector to search for bioproject id `PRJNA662453` to download the sequence list and sample information `metadata.csv`, save it to the current directory
## After downloading, check the files
ls -alsh ./
## 12K -rw-r--r-- 1 shpc_100419 shpc_100419 12K Jul 17 09:14 SraRunTable.txt
## 4.0K -rw-r--r-- 1 shpc_100419 shpc_100419 336 Jul 17 09:15 SRR_Acc_List.txtThen, we use <span>prefetch</span> to download —
# Create and switch to the working directory
mkdir -p ./qiime2_test/ALD_mice; cd ./qiime2_test/ALD_mice
# Download the required sequences from the sra database
## Choice 1: download the `.sra` files via 'prefetch' implemented in sra-tools
### First, equip sra-tools
micromamba install sra-tools
### Use the prefetch function to download sra files
nohup prefetch -l SRR_Acc_List.txt -O ./sra/ &Thus we begin the long wait for the good news (ku) (zao) (nan) (nai)
20 min…, ,ԾㅂԾ,,
40 min…( ̄、 ̄)
60 min…(ˉ▽ˉ;)
80 min passed, and only 3 sra files were barely downloaded…(⊙x⊙;)
prefetch: Calm down, it’s only 80 min 😛
In fact, from the SRA Run Selector webpage, we can see that there are only 28 samples in total, with a total data volume of about 312.2Mb, averaging about 11-12Mb per sample. Moreover, subsequent conversion using <span>fasterq-dump</span> to <span>.fastq</span> format, how can such a slow speed be tolerated (*  ̄︿ ̄)
If you want to improve download speed, Aspera is great, but unfortunately, the evil NCBI has migrated file storage to the cloud, making Aspera’s disk access-dependent algorithm useless, and the new version of <span>sra-tools</span> no longer supports Aspera download functionality.
By chance, I discovered another useful program <span>grabseqs</span>. <span>grabseqs</span> is an open-source tool based on Python developed by Louis J. Taylor and others at the University of Pennsylvania School of Medicine, first published in Bioinformatics in 2020, supporting data downloads and corresponding sample information from various platforms. <span>grabseqs</span> supports downloading data using SRA biosample, bioproject, or runs ID. This program relies on the <span>sra-tools</span> component, especially the <span>fasterq-dump</span> function, and compresses the obtained sequence files using <span>pigz</span>, so the final output is <span>.fastq.gz</span> instead of <span>.sra</span>. For the same reason, you must first download and install the <span>sra-tools</span> component before using <span>grabseqs</span>. Since we have already downloaded the <span>sra-tools</span> component when using <span>prefetch</span>, we will skip this step. Below we will try to use this tool to obtain <span>.sra</span> files. Since this tool is uploaded to the <span>python package index</span> and the <span>anaconda cloud repository</span>, readers can choose to configure it via <span>conda</span> or <span>pip</span> according to their preference.

# Choice 2: download the sequences via `grabseqs` function
# Install grabseqs
## Choice 1: via conda or mamba
conda install grabseqs
micromamba install grabseqs
## Choice 2: via pip
pip install grabseqs
grabseqs --help
# Attempt to download sequences based on grabseqs (taking one sample as an example)
## arguments: ## -l —— list the samples to be grabbed (but not download); ## -t —— set the number of threads; ## -r —— set the number of attempts to download
## View the sequences to be downloaded
grabseqs sra -l SRR12665664Then the problem arises —
---
Traceback (most recent call last): File "/home/micromamba/envs/qiime2-2022.2/bin/grabseqs", line 11, in <module> sys.exit(main()) File "/home/micromamba/envs/qiime2-2022.2/lib/python3.8/site-packages/grabseqslib/__init__.py", line 58, in main metadata_agg = process_sra(args, zip_func) File "/home/micromamba/envs/qiime2-2022.2/lib/python3.8/site-packages/grabseqslib/sra.py", line 31, in process_sra metadata_agg) File "/home/micromamba/envs/qiime2-2022.2/lib/python3.8/site-packages/grabseqslib/sra.py", line 97, in get_sra_acc_metadata run_col = lines[0].index("Run") ValueError: 'Run' is not in list
---What is going on? It seems there is an issue at the <span>get_sra_acc_metadata</span> step. Someone on GitHub has already speculated —
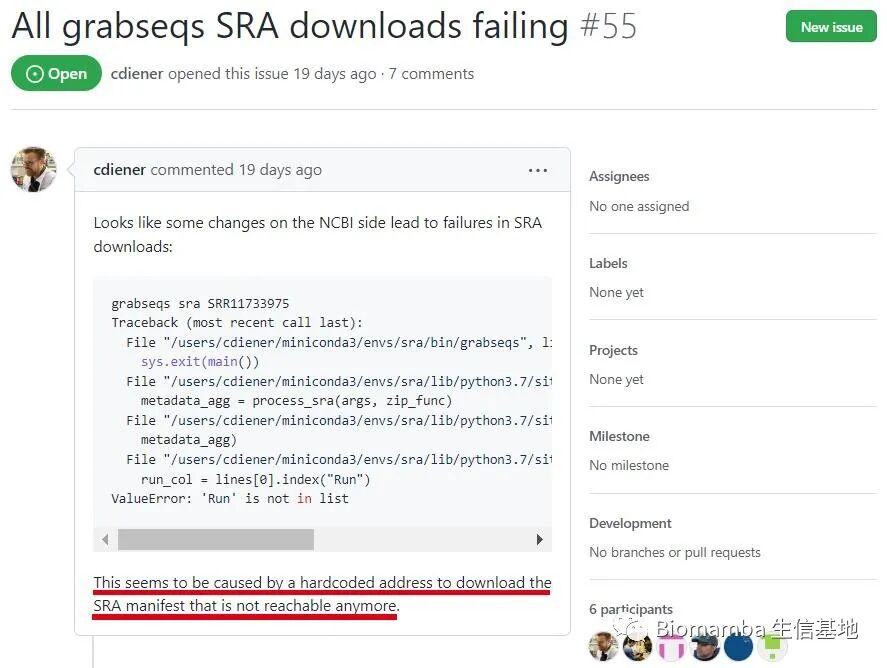
Alright, NCBI is going to be criticized again because of the retirement of the Trace Archive and the migration of data to the cloud, which has caused the original access address set by the program to become invalid, making it impossible to access the storage location of the required data, resulting in hard-coded addresses. This is exactly what the author speculated —
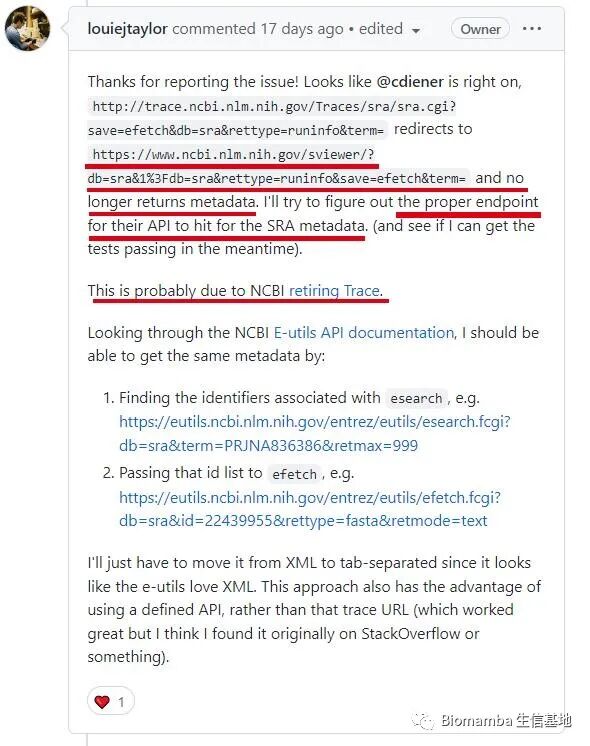
The solution is straightforward: change the access location of <span>grabseqs</span> to the new storage location —
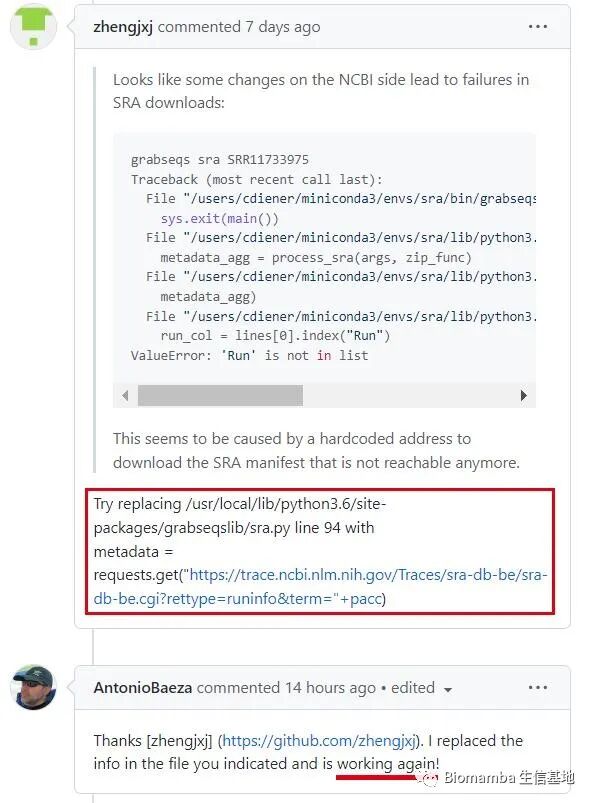
The original script’s preset access address looks like this, seemingly a comma-separated <span>.csv</span> file —
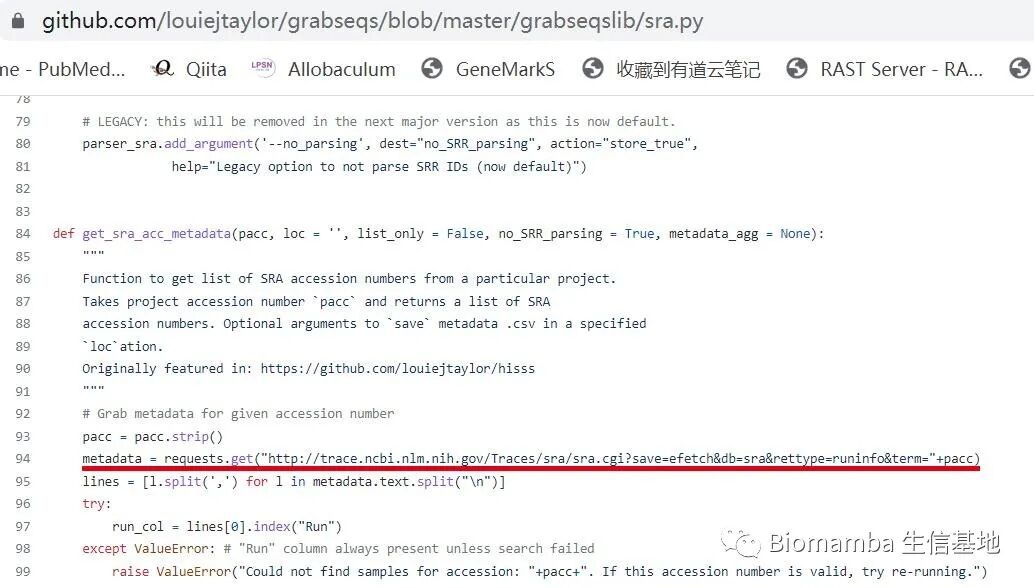
Before making changes, we can first observe what content is stored at this location through the browser —
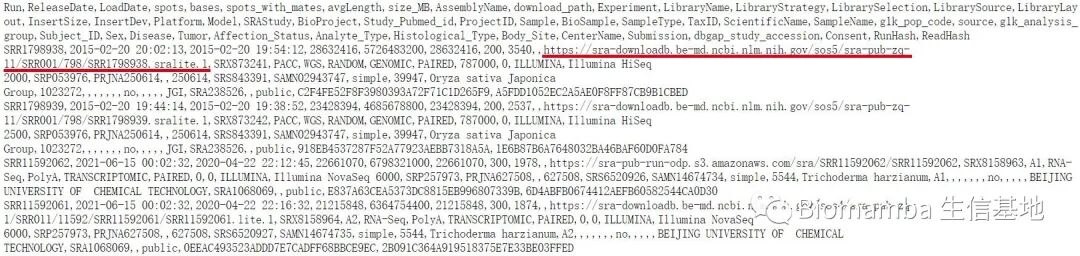
From the image, we see that this address stores relevant information about each sample sequence (the so-called <span>metadata</span>), including the actual storage location of the <span>.sra</span> files, separated by commas. From the storage address, it is clear that NCBI has indeed migrated a large amount of data to the cloud, no wonder Aspera is left in despair.
So the next steps are straightforward: first change the preset metadata storage location of <span>grabseqs</span>, and then attempt to download the sequence files.
# Change the access address set in sra.py to `https://trace.ncbi.nlm.nih.gov/sra-db-be/sra-db-be.cgi?rettype=runinfo&term="+pacc"
vi /home/micromamba/envs/qiime2-2022.2/lib/python3.8/grabseqslib/sra.py
# Use grabseqs to download sequences
time grabseqs sra SRR12665664 -t 12 -r 3The final standard output looks like this —

And the output files are the <span>.fastq.gz</span> files we have been eagerly anticipating (joyful news) —
ls -alsh ./
#4.0K drwxr-xr-x 4 user user 4.0K Jul 17 22:01 .
#4.0K drwxr-xr-x 3 user user 4.0K Jul 15 08:39 ..
#4.0K drwxr-xr-x 2 user user 4.0K Jul 17 18:57 raw_data
#4.0K drwxr-xr-x 2 user user 4.0K Jul 17 15:10 sra
# 12K -rw-r--r-- 1 user user 12K Jul 17 09:14 SraRunTable.txt
# 4.5M -rw-rw-r-- 1 user user 4.5M Jul 17 22:01 SRR12665664_1.fastq.gz
# 4.3M -rw-rw-r-- 1 user user 4.3M Jul 17 22:01 SRR12665664_2.fastq.gz
#4.0K -rw-r--r-- 1 user user 336 Jul 17 09:15 SRR_Acc_List.txt
# Use `grabseqs` to download all sequence files
## argument: `--custom_fqdump_args` —— parameters passed to `fasterq_dump`
## --split-3: splits paired-end sequencing into two files, but reads that are present in one end and not the other will be placed in a separate folder;
## --progress: outputs the program progress.
nohup time grabseqs sra PRJNA663684 -t 20 -r 5 -o ./ --custom_fqdump_args="--split-3 --progress"
# Download completed, taking about 78 min, while prefetch is still unhurried (╯‵□′)╯︵┻━┻
# Count the number of downloaded files
ls ./ | grep -c '_1.fastq'
ls ./ | grep -c '_2.fastq'
# 28 28, meaning a total of 28 forward sequences and 28 reverse sequences were downloaded.Thus, our sequence download work is complete. Next comes the data analysis work. Specific content will be shared in the next issue.

5. References
Bolyen E, Rideout JR, Dillon MR, et al. 2019. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nature Biotechnology 37: 852–857. https://doi.org/10.1038/s41587-019-0209-9
Qian M, Liu J, Zhao D, et al. Aryl Hydrocarbon Receptor Deficiency in Intestinal Epithelial Cells Aggravates Alcohol-Related Liver Disease. Cell Mol Gastroenterol Hepatol. 2022;13(1):233-256. https://doi.org/10.1016/j.jcmgh.2021.08.014
Mamba documentation. https://mamba.readthedocs.io/en/latest/index.html
Taylor LJ, Abbas A, Bushman FD. grabseqs: simple downloading of reads and metadata from multiple next-generation sequencing data repositories. Bioinformatics. 2020;36(11):3607-3609. https://doi.org/10.1093/bioinformatics/btaa167
Detailed usage of NCBI SRA database. https://www.cnblogs.com/lmt921108/p/7442699.html
Xu Zhougeng. Fastq-dump: A magical software. https://www.jianshu.com/p/a8d70b66794c

Previous Reviews
Introduction to Microbiomes:
Microbiome Research Techniques — Microbes Can Also Be Single-Cell
Overview of Research Ideas and Current Status of Microbes and Host Diseases
Literature Reading:
Literature Reading (1), Metabolic Networks of Human Gut Microbiota and DiseasesLiterature Reading (2), Changes in Gut Microbiota and Serum Metabolites Under Obesity and Weight Loss InterventionsLiterature Reading (16), Gut Microbiota & Kidney FailureLiterature Reading (17), Gut Microbiota & Metabolites & Hepatic CarcinogenesisLiterature Reading (20), Gut Microbiota of Adult T1D Patients and Host Blood Sugar
How to Contact Us

 Leave a message to receive materials and ready-to-use single-cell analysis images via WeChat ID [Biomamba_zhushou], making it convenient for everyone to communicate at any time. We have also built a group chat matrix, and everyone is welcome to join the discussion.After reading these articles, you can add tips for beginners in bioinformaticsWithout retrieval, there is no right to speak
Leave a message to receive materials and ready-to-use single-cell analysis images via WeChat ID [Biomamba_zhushou], making it convenient for everyone to communicate at any time. We have also built a group chat matrix, and everyone is welcome to join the discussion.After reading these articles, you can add tips for beginners in bioinformaticsWithout retrieval, there is no right to speak
Students who already have contact information for the bioinformatics basedo not need to add again

 Every like and view you give, I take seriously as a sign of appreciation
Every like and view you give, I take seriously as a sign of appreciation