Free amines easily react with NaOH or NaHCO3 under alkaline conditions controlled by a mixture of dioxane and water to produce N-tert-butoxycarbonylated amines via reaction with Boc2O. This is one of the commonly used methods for introducing Boc, which has the advantage of producing minimal interfering by-products that are easy to remove. For some nucleophilic amines, it is generally sufficient to directly react Boc anhydride in methanol without the need for additional bases, making it convenient to handle.
For amine derivatives that are sensitive to water, using Boc2O/TEA/MeOH or DMF at 40-50℃ is preferable, as these anhydrous conditions protect O17 labeled amino acids without losing O17 due to exchange with water. For sterically hindered amino acids, using Boc2O/Me4NOH.5H2O/CH3CN is very beneficial.
Aromatic amines, due to their weaker nucleophilicity, generally require the addition of a catalyst for the reaction. Additionally, for primary amines, the use of DMAP can facilitate the introduction of two Boc groups.
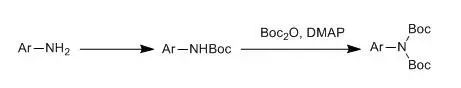
For amines with phenolic hydroxyl groups, the rate of Boc attachment to the phenolic hydroxyl is quite fast, hence generally lacks selectivity. When alcohol hydroxyl groups are present, if using DMAP as a catalyst, after a prolonged reaction time, the alcohol hydroxyl can also be substituted with Boc, therefore, it is advisable not to let the reaction proceed overnight.
Due to the formation of cyanate esters, sterically hindered amines often react with Boc2O to form ureas. This issue can be avoided by first reacting the amine with NaH or NaHMDS and then reacting with Boc2O.
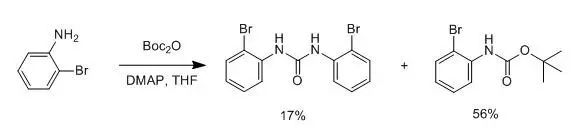
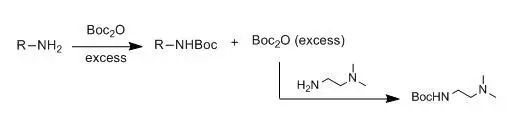
【How to Remove Excess Boc2O from the Reaction?】【What Common By-products Occur During Boc Removal?】【Should a Base be Added When Protecting an Amine with Boc? Can it be Used as a Solvent Without Column Purification and Without Adding a Base】
Reaction Examples
1. Amino Acid Boc Protection Example
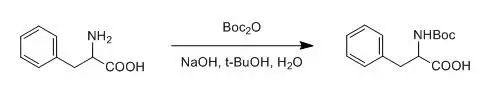
Oskar Keller, Walter E. Keller, Gert van Look et al., Org. Syn., 63, 160
A 4-L, four-necked, round-bottomed flask, equipped with an efficient stirrer, a dropping funnel, reflux condenser, and thermometer is charged with a solution of 44 g (1.1 mol) of sodium hydroxide in 1.1 L of water. Stirring is initiated and 165.2 g (1 mol) of L-phenylalanine is added at ambient temperature, and then diluted with 750 mL of tert-butyl alcohol. To the well-stirred, clear solution is added dropwise within 1 hr, 223 g (1 mol) of di-tert-butyl dicarbonate. A white precipitate appears during the addition of the di-tert-butyldicarbonate. After a short induction period, the temperature rises to about 30–35°C. The reaction is brought to completion by further stirring overnight at room temperature. At this time, the clear solution will have reached a pH of 7.5–8.5. The reaction mixture is extracted two times with 250 mL of pentane, and the organic phase is extracted three times with 100 mL of saturated aqueous sodium bicarbonate solution. The combined aqueous layers are acidified to pH 1–1.5 by careful addition of a solution of 224 g (1.65 mol) of potassium hydrogensulfate in 1.5 L of water. The acidification is accompanied by copious evolution of carbon dioxide. The turbid reaction mixture is then extracted with four 400-mL portions of ethyl ether. The combined organic layers are washed two times with 200 mL of water, dried over anhydrous sodium sulfate or magnesium sulfate, and filtered. The solvent is removed under reduced pressure using a rotary evaporator at a bath temperature not exceeding 30°C. The yellowish oil that remains is treated with 150 mL of hexane and allowed to stand overnight. Within 1 day the following portions of hexane are added with stirring to the partially crystallized product: 2 × 50 mL, 4 × 100 mL, and 1 × 200 mL. The solution is placed in a refrigerator overnight; the white precipitate is collected on a Büchner funnel and washed with cold pentane. The solid is dried under reduced pressure at ambient temperature to constant weight to give a first crop. The mother liquor is evaporated to dryness leaving a yellowish oil, which is treated in the same manner as described above, giving a second crop. The total yield of pure white N-tert-butoxycarbonyl-L-phenylalanine is 207–230 g (78–87%), mp 86–88°C, [α]D20 + 25.5° (ethanol c 1.0).
2. Direct Reaction of Boc Anhydride with Amine in Methanol
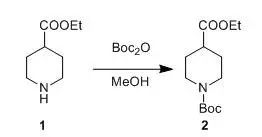
Boc2O (262 g, 1.2 mol) in MeOH (250 ml) was added to a solution of compound 1 (157.2 g, 1.0 mol) in MeOH (350 ml) at 10°C, and the resulting mixture was stirred at room temperature for 2 h. N1,N1-dimethylethane-1,2-diamine (26 g, 0.3 mol) was added and the mixture was stirred at room temperature for 15 min. The solvent was removed in vacuo, and the residue was dissolved with ethyl acetate (750 ml). The combined organics were washed with 1 N HCl (2 x 250 ml) and brine (2 x 250 ml), dried over sodium sulfate and filtered. The solvent was removed to give compound 2 (250 g, 96%), which was used directly in the next step.
3. Single Boc Protection Example for Aromatic Amines
Luo, Qun-Li; Liu, Zhi-Ying et al., J. Med. Chem., 2003, 46(13), 2631-2640
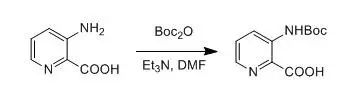
3-Aminopyridine-2-carboxylic acid (5.02 g, 36 mmol) was suspended in 60 mL of dry DMF, and Et3N (15.2 mL, 108 mmol) was added dropwise at room temperature. To the resulting brown solution was added Boc2O (11.80 g, 54 mmol). After being stirred for 10 min, the mixture was heated at 40-50 °C overnight. The reaction mixture was poured into water and was then extracted with EtOAc (2 X 50 mL). The aqueous phase was acidified to pH 4-5 with 2 M aqueous HCl and then extracted with CH2Cl2(3 X 50 mL). The combined organic phases were then processed in the usual way and chromatographed (13:1 CHCl3/MeOH) to yield the desired product (4.2 g, 49%).
4. Double Boc Protection Example for Aromatic Amines
Macleod, Calim; Mckieman, Gordon J et al., J. Org. Chem., 2003, 68(2), 387-401
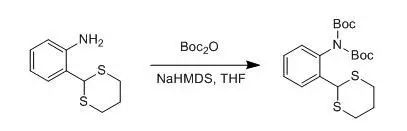
A solution of NaHMDS (22.0 mL, 22.0 mmol, 1 M in THF) was added to a solution of the amine (2.11 g, 10.0 mmol) and (Boc)2O (5.46 g, 25.0 mmol) in THF (50 mL) at 0°C under nitrogen. The reaction was allowed to warm to rt and stirred for 16 h. After this time, the reaction was poured into water, extracted into CH2Cl2(2 X 25 mL), washed with water (2 X 25 mL), dried over Na2SO4, and concentrated to yield a white-yellow solid. Recrystallization from petroleum ether (40-60 °C) gave the imide as needles (3.21 g, 7.80 mmol, 78%). Rf (hexane/ CH2Cl2 1:9, SiO2): 0.10. Mp:106-109 °C.
5. Amide Boc Protection Example
Lars G. J. Hammarström, Yanwen Fu et al., Org. Syn., 81, 213
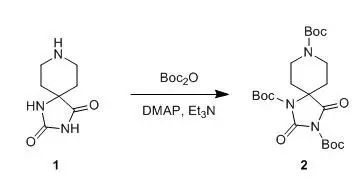
A 2000-mL, three-necked, round-bottomed flask equipped with an argon inlet adapter, glass stopper, and an overhead mechanical stirrer is charged with a suspension of the hydantoin 1 (26.0 g, 154 mmol) in 1000 mL of 1,2-dimethoxyethane. Triethylamine (15.7 g, 154 mmol) is added in one portion, and the resulting white suspension is stirred for 30 min. Di-tert-butyl dicarbonate (168.0 g, 770 mmol) is then added by pipette, followed by 4-dimethylaminopyridine (DMAP) (0.2 g, 1.5 mmol). Six additional 0.2 g-portions of DMAP are added at 12 hr intervals during the course of the reaction. The reaction mixture is stirred vigorously for a total of 72 hr, and the resulting light yellow solid is then collected in a Büchner funnel using suction filtration. The filtrate is concentrated to a volume of 60 mL by rotary evaporation, and the resulting solution is cooled to 15°C. The precipitate which appears is collected using suction filtration, added to the first crop, and the combined solids are dissolved in 500 mL of chloroform. This solution is washed with three 200-mL portions of 1.0N HCl, and the combined aqueous phases are extracted with 100 mL of chloroform. The combined organic layers are washed with 100 mL of saturated aq NaHCO3 solution and 100 mL of brine, dried over anhydrous MgSO4, filtered, and concentrated by rotary evaporation. The resulting solid is dried at room temperature at 0.01 mm for 24 hr. The resulting finely ground light yellow solid is suspended in 400 mL of diethyl ether in a 1000-mL, round-bottomed flask equipped with a magnetic stir bar, stirred for 2 hr, and filtered on a Büchner funnel washing with four 50-mL portions of diethyl ether. The product is dried under vacuum (85°C; 0.5 mm) for 24 hr to give 60.0–65.3 g (83-90%) of 2 as an ivory-colored solid.
6. Indole Boc Protection Example
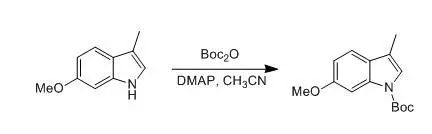
G. Tong; P. Ruiyan et al., J. Org.Chem., 1997, 26, 9298
To a solution of 6-methoxy-3-methylindole (5.0 g, 31 mmol) in distilled acetonitrile (150 mL) were added di-tert butyl dicarbonate (7.44 g, 34.1 mmol) and DMAP (0.195 g, 1.6 mmol). The reaction mixture was stirred at rt for 12 h. The solvent was removed under reduced pressure. The residue was dissolved in CH2Cl2(100 mL) and washed with an aqueous solution of 1 N HCl (2 x 50 mL). The aqueous layer was extracted with CH2Cl2(3 x 30 mL). The combined organic layers were dried (K2CO3). After removal of solvent under reduced pressure, the residue was solidified to afford the product (8.12 g, 99%) as a yellow solid: mp 45-46 °C.
Boc Deprotection
Boc is more sensitive to acid than Cbz, and the acidolysis products are isobutylene and CO2 (see below). In the liquid-phase peptide synthesis, Boc removal is generally done using TFA or 50% TFA (TFA:CH2Cl2= 1:1, v/v). In solid-phase peptide synthesis, due to some side reactions caused by TFA (such as the introduction of trifluoroacetyl groups on the resulting amines), 1-2M HCl/organic solvent is often used. Generally, HCl/dioxane is more common.
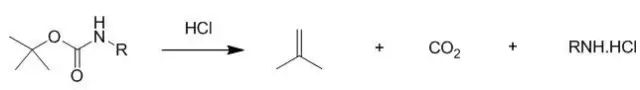
Using methanol as a solvent, the combination of HCl/EtOAc allows TBDMS and TBDPS esters [1] as well as tert-butyl esters and non-phenolic esters to not be cleaved during Boc removal, except for S-Boc [2]. However, when simultaneously removing Boc and tert-butyl esters in the same molecule, or when there is a free carboxylic acid group present in the molecule, it is critical not to use HCl/MeOH, as this can convert the carboxylic acid into a methyl ester. Meanwhile, AcCl/MeOH is a convenient method for generating anhydrous HCl in methanol. These conditions can also be used to prepare esters from carboxylic acids and form hydrochloride salts of amines[3].
Under neutral anhydrous conditions, Me3SiI in CHCl3 or CH3CN can remove Boc while also cleaving carbamates, esters, ethers, and ketones. By controlling the conditions, some selectivity can be achieved[4].
When certain functional groups in the molecule can react with the by-product tert-butyl carbocation under acidic conditions, thiophenols(such as phenyl thiol) need to be added to remove the tert-butyl carbocation, which can prevent the alkylation during Boc removal of methionine and tryptophan[5]. Other scavengers can also be used, such as benzyl ether, phenyl thioether, tolyl thiol, tolyl phenol, and dimethyl sulfide[6]. During the Boc removal process, TBDPS[7] and TBDMS[8] groups are stable towards CF3COOH (in the presence of TBS, using relatively dilute 10-20%TFA). In the presence of primary amine derivatives, ZnBr2/CH2Cl2 can selectively remove the Boc from secondary amines.
1. F. Cavelier, C.Enjabal., Tetrahedron Lett., 1996, 37, 5131
2. F. S. Gibson, S. C. Bergmeier, H. Rapoport., J. Org. Chem., 1994, 59, 3216
3. A. Nudelman, Y. Bechor et al., Synth. Commun., 1998, 28, 471
4. R. S. Lott, V. S. Chauhan et al., J. Chem. Soc. Chem. Commun., 1979, 495; G. A. Olah, S. C. Narang., Tetrahedron., 1982, 38, 2225
5. R. A. T. M. van Benthem, H. Hiemstra et al., I J. P. P. Res,12,258(1978) 7, 6083
6. M. Bodanszky, A. Bodanszky., Int. J. Pept. Protein Res., 1984, 23, 565; Y. Masui, N. Chino et al., Bull. Chem. Soc. Jpn., 1980, 53,464
7. P. A. Jacobi, S. Murphree et al., J. Org. Chem., 1996, 61, 2413
8. J. Deng, Y. Hamada et al., J. Am. Chem. Soc., 1995, 117, 7824
9. S. C. Nigam, A. Mann et al., Synth. Commun.,1989, 19, 3139
Reaction Examples
1. Deprotection of Boc under Neutral Conditions with TMSOTf
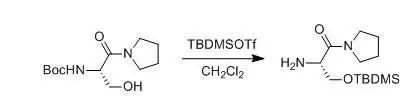
Gilbertson, Scott R; Chang, Cheng-Wei et al., J.Org. Chem., 1998, 63(23), 8424-8431
To a solution containing 2 (1.0 g, 3.9 mmol) in 30 mL of dry CH2Cl2 was slowly added TBDMSOTf (0.9 mL, 4.1 mmol). After stirring the reaction mixture for 6 h, the solvent was evaporated, and the crude product (0.8 g, 75%) was obtained, which was used directly in the next step.
2. Deprotection of Boc under Neutral Conditions with TMSOTf-2,6-lutidine
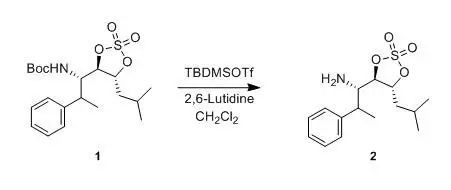
Kemp, Scott J; Bao, Jiaming et al J. Org.Chem., 1996, 61(20), 7162-7167
To a stirring solution of compound 1 (800 mg, 2.0 mmol) and 2,6-lutidine (0.4463 ml, 4.0 mmol) in CH2Cl2(6 mL) was added tert-butyldimethylsilyl triflate (0.690 ml, 3.0 mmol) dropwise over 5 min. After 20 min, saturated NH4Cl (10 mL) was added. The mixture was stirred and separated, and the aqueous layer was extracted with Et2O (3 x 15 mL). The combined organic layers were washed with water (2 x 10 mL) and saturated NaCl (10 mL), dried (MgSO4), and concentrated to give the crude silyl carbamate, which was dissolved in THF (10 mL) and cooled to 0°C. A 1.0 M solution of TBAF in THF (2 mL, 2 mmol) was added over 5 min, and then the solution was stirred at 0°C for 1 h. The solution was concentrated and chromatographed (95:5 CH2Cl2-methanol) through a small plug of silica to give compound2 (882 mg, 75%) as a clear oil.
3. TFA Deprotection of Boc
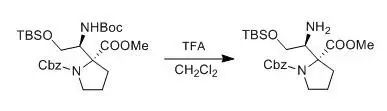
M. Alberto; A. Eduardo et al., J. Org. Chem., 2004, 21,7004
To a solution of the β-aminoester (0.2 mmol) in CH2Cl2 (3 mL), cooled to 0°C was added TFA (1 mL). After the consumption of the starting material (45 min, monitored by TLC), the mixture was evaporated and then saturated aqueous NaHCO3 was added. The aqueous layer was extracted twice with CH2Cl2 (15 mL), and the organic layer washed with brine and dried over anhydrous Na2SO4. The solvent was removed under vacuum, to afford the amine, which were employed without further purification to prepare the Mosher’s diastereoisomeric amides.
4. Deprotection of Boc with HCl-THF
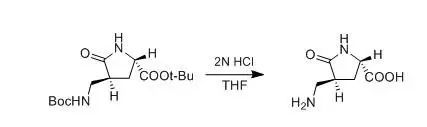
J. Wehbe et al., Tetrahedron: Asymmetry, 2004, 15, 851
To the Boc protected amine (0.06 g, 0.17 mmol) dissolved in THF (1 mL) was added 2M HCl (1 mL, 2 mmol) and the mixture stirred 2 h at room temperature. After evaporation of the solvent, the product was extracted into EtOAc (3.5 mL). The organic layer was dried and evaporated under vacuum to afford 17b in 95% yield as a white solid.
5. Deprotection of Boc in the Presence of Tert-Butyl Ester Example 1
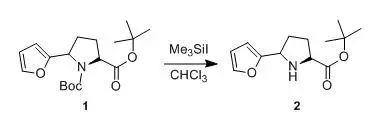
US5610144
1.77 ml of Me3SiI are added dropwise at room temperature in the vicinity of 25°C to a solution of 3.8 g of compound 1 in 50 ml of CHCl3. Stirring is continued for 30 min, then 20 ml of water are added. The aqueous phase is separated, then extracted with CHCl3 (2x 20 ml). The organic phases are combined, washed successively with a saturated aqueous Na2CO3 (30 ml) and water (2 x 30 ml), then dried over MgSO4 and concentrated to dryness under reduced pressure at 40°C. The mixture of the two diastereoisomers obtained is separated by chromatography on silica (eluent: ethyl acetate/cyclohexane = 1/4). The fractions containing the expected product are combined and concentrated to dryness under reduced pressure at 40°C to give compound 2 (0.5 g), as a yellow-orange oil, used as it is in subsequent syntheses.
6. Deprotection of Boc in the Presence of Tert-Butyl Ester Example 2
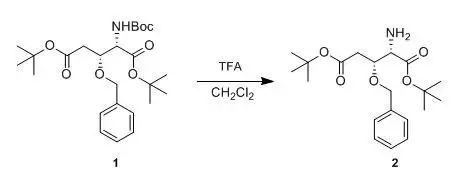
WO20040106286
To a solution of compound 1 (149 mg, 0.33 mmol) in CH2Cl2 (2 ml), TFA (1 ml) was added at 0°C and the mixture was stirred for 1 h at 0°C. Saturated aqueous Na2CO3 was added and the mixture was extracted with CHCl3. The extract was purified by silica gel column chromatography to obtain compound 2 (92 mg, 79%).
7. Deprotection of Boc with Oxalyl Chloride
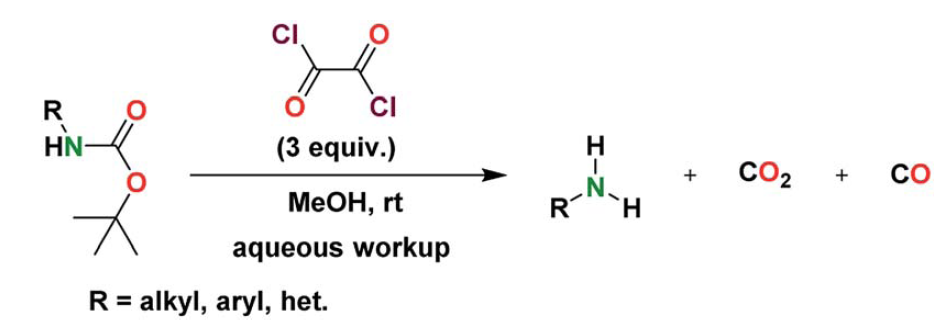
General Procedure for the N-Boc Deprotection
In a dry 25 mL round bottom flask equipped with a stirring bar,
the starting material (50 mg) was dissolved in MeOH (3 mL) and
allowed to stir at room temperature for 5 min. Oxalyl chloride (3
equiv.) was then added to the solution (via syringe or micropipette)
directly into the reaction solvent mixture. Sputtering and an increase in temperature of the reaction mixture were observed immediately upon addition of the oxalyl chloride. The reaction mixture was allowed to stir for up to 4 h depending on the starting material. The reaction was monitored via TLC. Upon complete conversion of the N-Boc-protected amine, deionized water (5 mL) was added to the flask slowly. The crude material was subsequently extracted with dichloromethane (5
mL) and washed with deionized water twice (2 x 5 mL). The organic layer was dried over anhydrous MgSO4, filtered, and concentrated under low vacuum on a rotary evaporator. Some products required further purification via flash column chromatography to yield the pure deprotected amine.
【RSC Adv., 2020, 10, 24017–24026】
This article is not original content; copyright belongs to the original author.
