1. Introduction
Acyclic compounds are important intermediates in the synthesis of many pharmaceuticals, and the synthesis of acyclic compounds is a very important part of organic synthesis. They are generally prepared by several methods:
1. Dehydration of amides
2. Reaction of aliphatic halides or sulfonate esters
3. Cyanation of aromatic halides
4. Other conversions from hydroxyl or oxime to nitriles
2. Dehydration Reaction of Amides
The dehydration reaction of amides can proceed in the presence of dehydrating agents such as P2O5, POCl3, SOCl2, and PCl5 to generate nitriles, making it one of the laboratory methods for synthesizing nitriles.
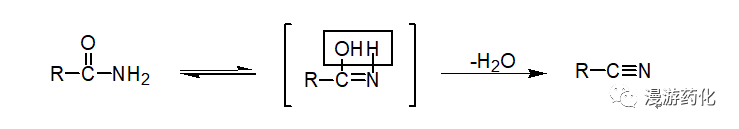
Heating a mixture of the amide and P2O5 can yield a good yield of the resulting nitrile upon distillation. SOCl2 is most suitable for handling higher amides because the byproducts are all gases, which are easy to remove, thus reducing the difficulty of purifying the nitrile.
At the same time, these dehydrating agents generally react under acidic conditions, which are impractical for acid-sensitive substrates. Therefore, many milder methods for the dehydration of amides have been developed, such as: Burgess reagent [Et3N+SO2N–COOMe], trifluoroacetic anhydride (TFAA) – triethylamine, (COCl)2-NEt3-DMSO, etc., which can react under low temperature and nearly neutral conditions. Additionally, methane sulfonyl chloride (CH3SO2Cl) and titanium tetrachloride (TiCl4) are also used.
2.1 Example of Dehydration Using P2O5
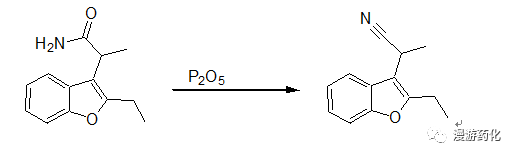
A solution of 35g (0.16 mol) of 2-(2-ethyl-3-benzofuranyl)-propionamide in 500mL of toluene was refluxed for 18 hours in the presence of P2O5. The organic phase was decanted off and the residue was carefully decomposed with ice-water and extracted with ether. The organic phase was washed with water, dried over sodium sulfate, and added to the toluenic phase. The solvent was evaporated off under reduced pressure, and the residue was fractionated to give 23.8g of 2-(2-ethyl-3-benzofuranyl)-propionitrile (yield 74.4%, boiling point: 105°C at 0.2 mmHg).
Reference: US4124710 A1 (1978/11/07)
2.2 Example of Dehydration Using POCl3
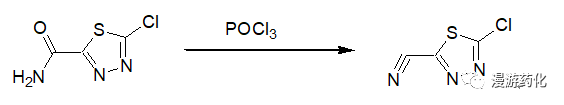
A mixture of 2-chloro-1,3,4-thiadiazole-5-carboxamide (1.4 g) in 17 ml of POCl3 is heated at reflux for 18 hours. The reaction mixture is concentrated, and the residue is suspended in 25 ml of ethyl acetate. The suspension is cooled in an ice bath and neutralized with saturated, aqueous NaHCO3 (to pH 7). The phases are separated, and the aqueous phase is extracted with 20 ml of ethyl acetate. The combined organic phases are dried over MgSO4, filtered, and concentrated. The residue is purified by column chromatography (using 30 percent ethyl acetate / hexane as eluent) to afford 0.832 g of 2-cyano-5-chloro-1,3,4-thiadiazole. MP: 65-67°C
Reference: Patent; EP883611 B1 (2002/07/31)
2.3 Example of Dehydration Using SOCl2
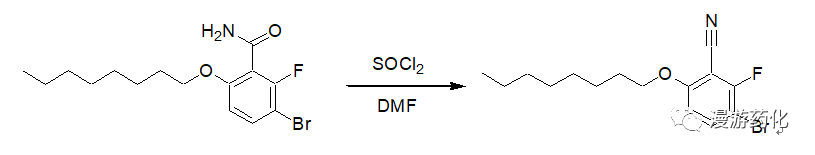
A solution of thionyl chloride (7.70 g, 0.065 mol) in dry DMF (10 ml) was added dropwise to a stirred solution of compound 13 (4.20 g, 0.013 mol) in dry DMF (25 ml) at room temperature. The stirred mixture was heated at 120°C for 3h and poured into ice–water. The product was extracted into ether (twice), and the combined ethereal extracts were washed with water, saturated sodium bicarbonate solution, water, and dried (MgSO4). The solvent was removed in vacuo, and the residue was purified by column chromatography (silica gel–light petroleum (bp 40–60°C) with the gradual introduction of dichloromethane) to yield a colorless solid. Yield 2.88 g (68%);
Reference: J. Chem. Soc.,Perkin Trans. 1, 1998, 3479–3484
2.4 Example of Dehydration Using PCl5
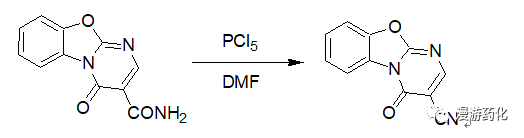
4-Oxo-4H-9-oxa-1,4a-diaza-fluorene-3-carboxylic acid amide (4.58 g, 20 mmol) was suspended in 150 ml of anhydrous DMF, PCl5 (5.0 g, 24 mmol) was added, and the mixture was stirred for 2 h at 40-50°C. The reaction mixture was poured into 600 ml ice-water to yield a solid, which was collected by filtration. The solid was washed thoroughly (first with saturated aqueous NaHCO3, then with water) and dried to give 4-oxo-4H-9-oxa-1,4a-diaza-fluorene-3-carbonitrile.
Ref: J. Med. Chem. 1983, 26, 608-611
2.5 Example of Dehydration Using Burgess Reagent
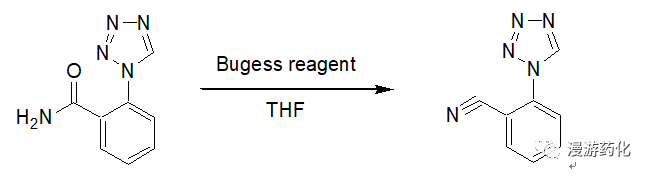
To a solution of 2-tetrazol-1-yl-benzamide (1.5 g, 7.9 mmol) in tetrahydrofuran (50 ml) was added Et3N+SO2N–COOMe (2.8 g, 11.8 mmol) in three portions over 1.5 h. Water was added, and the reaction mixture was extracted with ethyl acetate. The combined organic layers were washed with brine and water. After drying and filtration, the solvent was evaporated to give 2-tetrazol-1-yl-benzonitrile.
Reference: J. Med.Chem. 47, 12, 2004, 2995-3008.
Preparation of Burgess reagent:
A mixture of anhydrous methanol 19.2g (0.6 mol) and anhydrous benzene 40mL was added dropwise over 30-40 minutes to a mixture of ClSO2NCO 85g (52.3 mL, 0.6 mol) and anhydrous benzene 200mL, controlling the temperature at 10-15°C. After completion, stir at room temperature for 2 hours. Then add 1000mL of anhydrous benzene for dilution, and carefully add a mixture of 190mL of anhydrous triethylamine and 250mL of anhydrous benzene, controlling the temperature at 10-15°C, and complete the addition in about 40 minutes. After completion, stir at room temperature for 2 hours, a large amount of solid precipitates. After the reaction, filter, wash the solid with 200mL of anhydrous benzene and 200mL of anhydrous THF, then concentrate the filtrate (controlling the temperature <30°C), dissolve in anhydrous THF, and recrystallize to obtain 123g, yield 86%. Note: The entire operation temperature should be below 30°C.
2.6 Example of Dehydration Using TFAA-NEt3
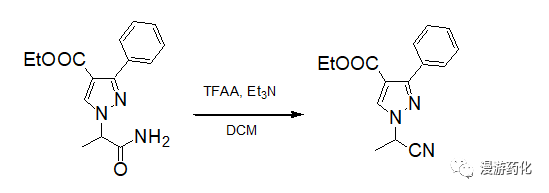
To a mixture of compound amide (287 mg, 1 mmol), Et3N (470 mg, 4.5 mmol) in anhydrous DCM (4 mL) was added TFAA (0.44 g, 2 mmol) at 0°C with stirring. The resulting mixture was warmed to room temperature and stirred for 12h. The reaction was monitored by TLC (Hexane:AcOEt = 1:1) until its completion. The organic layer was washed with brine and water, dried, and concentrated to give the desired product (~80% yield).
2.7 Example of Dehydration Using (COCl)2-NEt3-DMSO
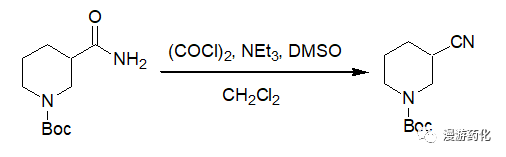
A solution of (COCl)2 (67 μL, 0.77 mmol) in CH2Cl2 (0.5 mL) was added to the solution of 3-carbamoyl-piperidine-1-carboxylic acid tert-butyl ester (142.0 mmol) and DMSO (78 μL, 1.1 mol) in CH2Cl2 (1.5 mL) at -78°C. After stirring for 15 min at -78°C, Et3N (0.23 mL, 1.65 mmol) was added dropwise to the mixture. After the reaction mixture was stirred for 15 min at -78°C, the mixture was quenched by addition of water (5 mL). After this mixture was warmed to room temperature, the aqueous layer was extracted with EtOAc (3×10 mL). The combined organic layers were washed with brine, dried, and filtered. Concentration after filtration in vacuo followed by purification by column gave 3-cyano-piperidine-1-carboxylic acid tert-butyl ester (123.3 mg, 93%).
Reference: T. L. 38, 12, 1997, 2099-2102
2.8 Example of Dehydration Using Methanesulfonyl Chloride (CH3SO2Cl)
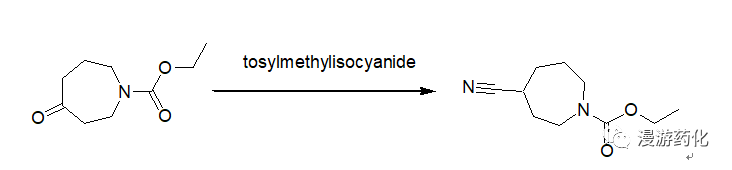
6-(3-Methoxy-2-propyl-phenyl)-hexanoic acid amide (7.2g, 27.2 mmol) was cooled to 0°C and added methane-sulfonyl chloride (18.5 mL, 239 mmol) dropwise over 5 min. The mixture was stirred overnight while slowly warming to 25°C. The reaction mixture was then poured into 3 volumes of ice water. The aqueous mixture was repeatedly extracted with ethyl acetate. The combined organic extracts were washed with dilute HCl and brine, then dried over MgSO4. After evaporation of the solvent, a brown oily residue was obtained. The crude nitrile was purified by bulb-to-bulb distillation (bp 133-137°C (0.02mmHg)), which was pure enough for further transformation (5.50 g, 83%).
Reference: J. Med. Chem. 1988, 31, 172-175
2.9 Example of Dehydration Using TiCl4
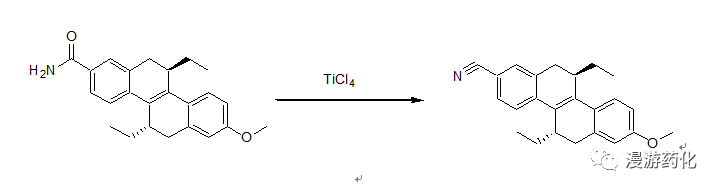
To a solution of CCl4 (110 μL, 1.17 mmol) and THF (6 mL) at 0°C was added TiCl4 (58 μL, 0.52 mmol). After 5 min, 5,11-diethyl-8-methoxy-5,6,11,12-tetrahydro-chrysene-2-carboxylic acid amide (47 mg, 0.13 mmol) in THF (14 mL) and Et3N (72 μL, 0.52 mmol) was added to this yellow heterogeneous solution, and stirring was continued at room temperature until no starting material remained. Diethyl ether and water were added, and the organic layer was washed with brine, dried over MgSO4, and concentrated. Repeated recrystallization from diethyl ether gave 5,11-diethyl-8-methoxy-5,6,11,12-tetrahydro-chrysene-2-carbonitrile (45 mg, 99%).
Reference: J. Org. Chem. 1992, 1262-1271
2.10 Dehydration of Tert-Butyl Amide to Nitrile
Tert-butyl amide can also serve as a substitute for primary amides, and can be dehydrated to nitriles under the action of thionyl chloride, phosphorus oxychloride, or oxalyl chloride. Therefore, it is sometimes a good method to convert the corresponding tert-butyl amide to nitrile when it is not easy to prepare primary amides.
2.10.1 Example of Dehydration of Tert-Butyl Amide to Nitrile Example 1
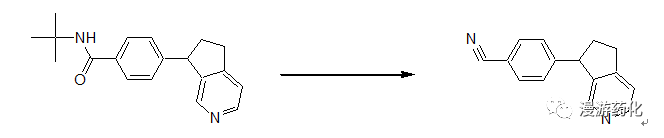
A solution of 1.240 mmol of N-tert-butyl-4-(6,7-dihydro-5H-[2]pyridin-7-yl)benzamide and 1.0 ml of thionyl chloride in 30 ml of chloroform is stirred under reflux for 6 hours. The reaction mixture is cooled to room temperature and evaporated. The residue is taken up in dichloromethane and mixed with saturated aqueous sodium bicarbonate solution. The organic phase is separated and the aqueous phase is extracted with dichloromethane (2x). The combined organic phases are dried with sodium sulfate and concentrated. The residue is dissolved in diethyl ether and the title compound is converted into the hydrochloride salt by adding ethereal HCl solution (2N). The solid is stirred in diethyl ether/acetone (1: 1), filtered, and dried. The title compound is obtained as a dark grey solid. Rf (free base) = 0.36 (EtOAc)
Reference: WO2005/118540
2.10.2 Example of Dehydration of Tert-Butyl Amide to Nitrile Example 2

A 5 L round bottom flask was charged with N,N’-di-tert-butyl-5-(2,3-difluoro-6-nitro-phenoxy)-isophthalamide (21; 564 g) and 1.3 L of phosphorus oxychloride. The mixture was heated to between 90°C ~ 100°C for 2 h, after which approximately 1/2 of the POCl3 was removed by distillation. Toluene was added (1 L) and additional liquid was distilled. After cooling the mixture overnight, a crude was obtained by filtration. Additional material was obtained by recovery from the mother liquid. The combined solids were stirred in MeOH (0.7L) for between 1 and 3 h, filtered and dried in a vacuum oven between 50~80°C at 25 Torr with a nitrogen bleed to afford 339 g of 22 (90 percent theory).
Reference: US2005/234236
2.10.3 Example of Dehydration of Tert-Butyl Amide to Nitrile Example 3
At ice-water bath, oxalyl chloride (0.345 ml) was added dropwise to a solution of 1.0 g of ethyl 1-{4-[2-(t-butylaminocarbonyl)phenyl]phenyl}methyl-4-(1-hydroxy-1-methylethyl)-2-propylimidazole-5-carboxylate in 10 ml of methylene chloride. The mixture was stirred at the same temperature for 2 hours. The reaction mixture was diluted with an aqueous solution of sodium bicarbonate and ethyl acetate, and the ethyl acetate layer was separated, dried over anhydrous magnesium sulfate, and concentrated by evaporation under reduced pressure. The residue was purified by silica gel column chromatography, using 1:1 EtOAc/hex (v/v) as the eluent, to give 0.69 g of the title compound as crystals.
Reference: US5616599
3. Nucleophilic Substitution Reaction of Aliphatic Halides or Sulfonate Esters with Metal Cyanides
The nucleophilic substitution reaction in aliphatic systems is one of the most noted unit reactions by organic chemists, where the nucleophilic substitution of aliphatic halides or sulfonate esters with metal cyanides has been widely applied:
In the conversion synthesis process, the most useful substrates are those with reactive activity in direct substitution mechanisms, i.e., primary and unhindered secondary aliphatic halides or sulfonate esters. The tendency for elimination reactions in tertiary alkyl systems is quite significant, thus limiting the application of nucleophilic substitution reactions in conversion synthesis involving these systems. Sometimes, when the reactivity of non-iodinated halides is insufficient, KI or NaI needs to be added to increase the reactivity of the halides, or if oxygen ion complexing agents, such as 18-crown-6, are present; many literatures have reported achieving this substitution using phase transfer catalysis methods.
Aliphatic halides can be prepared from the corresponding alcohols through halogenation, while sulfonate esters can be obtained from the corresponding alcohols by reacting with methanesulfonyl chloride or p-toluenesulfonyl chloride.
3.1 Example of Cyanide Substitution Reaction of Alkyl Halides
To a stirring solution of sodium cyanide (1.62 g, 33 mmol) and potassium iodide (66 mg, 0.4 mmol) in dimethyl sulfoxide (20 ml) at 40°C, was slowly added 1-bromo-2-ethylbutane over 30 min. The reaction mixture was stirred at 80°C for 12 h, then at 110°C for 4 h. The reaction mixture was cooled and partitioned between Et2O and water. The organic layer was washed with brine, dried over Na2SO4, filtered, and concentrated in vacuo to yield 2.9 g (87 percent yield) of 1-cyano-2-ethylbutane as an amber oil.
1H NMR (300 MHz, CDCl3): d 2.34 (d, 2 H), 1.58 (m, 1 H), 1.46 (m, 4 H), 0.92 (t, 6 H).
Reference:: Bioorg. Med.Chem. 11, 18, 2003, 4093-4102.
3.2 Example of Cyanide Substitution Reaction of Sulfonate Esters
A mixture of 1-[(4-butylphenyl)methyl]-3-(4-chloro-2-methylphenyl)-1-[6-[(methylsulfonyl)oxyl]-hexyl]urea (2.0 g) in a few mL of DMF was added to a cooled stirring suspension of anhydrous sodium cyanide (0.50 g) in 3 mL of dry DMSO. The mixture was heated overnight at 80°C. Then it was poured into water (100 mL), and the product was extracted with dichloromethane. The combined extracts were washed with water, dried (MgSO4), and concentrated to give 1-[(4-butylphenyl)methyl]-3-(4-chloro-2-methylphenyl)-1-(6-cyanohexyl)urea.
Ref: US 4623662 A1
4. Conversion of Hydroxyl to Nitriles Using TMSCN
For hydroxyls at the benzylic position of aryls, TMSCN can be directly converted to nitriles, and the success of the reaction depends on whether there are alkyl hydrogen protons on the neighboring carbon.
4.1 Example of Cyanation Reaction of Diphenylmethanol Using TMSCN
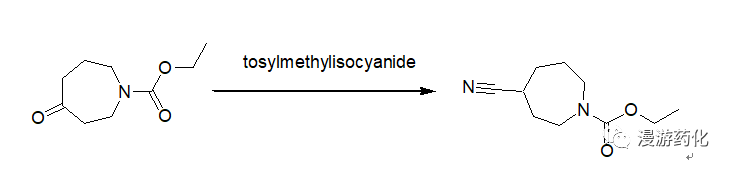
Thionyl chloride (50 ml) was added to bis(4-fluorophenyl)methanol (24.2 g) at 0°C and after stirring for 30 min, the mixture was poured into 2N hydrochloric acid (500 ml). The mixture was extracted with ethyl acetate and the organic layer was dried over calcium chloride and concentrated under reduced pressure. The resulting residue was dissolved in dichloromethane (200 ml) and after addition of trimethylsilylcyanide (16.4 ml), titanium tetrachloride (13.4 ml) was added dropwise at 0°C. The mixture was stirred for 50 min. Methanol (5 ml) was added to the reaction mixture, and the mixture was poured into saturated aqueous sodium hydrogen carbonate. The mixture was extracted with ethyl acetate and washed with saturated brine. The organic layer was dried over anhydrous magnesium sulfate and concentrated under reduced pressure to give the title compound (21.8 g) oil.
Ref: EP1219294 A1 (2002/07/03)
4.2 Example of Cyanation Reaction of Monophenylmethanol Using TMSCN
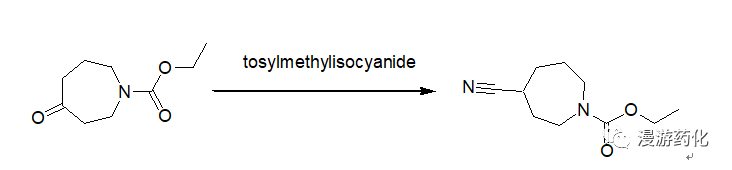
To a solution of 4-(1-cyclohexyl-3-ethyl-1H-indazol-6-yl)-4-hydroxy-cyclohexanecarboxylic acid ethyl ester (13.5 g, 33.9 mmol) in CH2Cl2 (135 mL) cooled to 0 °C was added trimethylsilyl cyanide (22.6 mL, 169 mmol) followed by a slow addition of SnCl4 (13.6 mL of a 1.0 M solution in CH2Cl2, 13.6 mmol). The reaction mixture was allowed to warm to room temperature overnight. K2CO3 (18.7 g, 136 mmol) and KF·2H2O (12.8 g, 136 mmol) were added, followed by dropwise addition of H2O (4.30 mL, 239 mmol). The reaction mixture was stirred vigorously for 90 min, after which silica gel (25 g) was added. The mixture was filtered and washed thoroughly with CH2Cl2. The filtrate was washed with saturated aqueous NaHCO3 (250 mL), dried over MgSO4, filtered, and concentrated to yield 13.2 g oily product of (96% recovery) cyano-4-(1-cyclohexyl-3-ethyl-1H-indazol-6-yl)cyclohexanecarboxylic acid ethyl ester as a mixture of diastereoisomers. For characterization purposes, a sample of each diastereoisomer was obtained by chromatographic purification on silica gel eluting with 4:1 hexanes/EtOAc.
Reference:: Organic Process Research & Development 2001, 5, 587-592
5. Direct Conversion of Ketones to Cyanides Using TosMIC
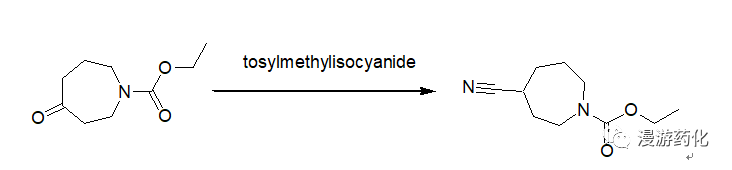
To a 250 mL round-bottomed flask equipped with a condenser and nitrogen inlet were added 4.34 g (23.49 mmol) N-carboethoxyperhydroazepin-4-one (prepared according to the procedure given by Z. G. Finney and T. N. Riley, J. Med. Chem., 23, 895, 1980), 10.53 g (54.02 mmol) tosylmethylisocyanide and 117 mL 1,2-dimethoxyethane. The solution was cooled to 0°C and 2.48 mL (54.02 mmol) ethanol and 9.21 g (82.2 mmol) potassium t-butoxide were added. The mixture was heated at 60°C for 18 hours, cooled and concentrated. The residue was taken up in ethyl acetate, washed with brine, dried over sodium sulfate and concentrated to give an oil. The oil was purified by chromatography on silica gel using hexane/ethyl acetate as eluent to afford 4.6 g (100 percent) of oil.
Reference: 72800; Patent; Pfizer Inc.; Publ.: US5373003 A1 (1994/12/13)
6. Conversion of Ketones to Cyanides Using 2,4,6-Triisopropylbenzenesulfonohydrazide-KCN
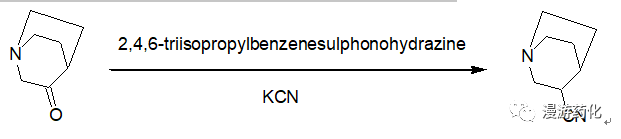
3-Quinuclidinone (24.2 g, 0.19 mol) and 2,4,6-triisopropylbenzenesulphonohydrazide (72g, 0.24 mol) were stirred together in anhydrous MeOH (250 mL) for 3 h. Potassium cyanide (33.8 g, 0.51 mol) was added, and the mixture was heated under reflux for 5 h. The residue after evaporation of the solvent was partitioned between water and CH2Cl2. The organic phase was dried and evaporated, and the residue was fractionally distilled under reduced pressure to give 3-cyanoquinuclidine (32, 6.1 g).
Reference:: J.Med. Chem. 1990, 33, 1128-1138
7. Aromatic Halides Under Metal Catalysis Nitrilation Reaction
Aromatic nitriles play a very important role in organic synthesis, especially in the applications of dyes, herbicides, agricultural chemicals, pharmaceuticals, and natural products. Traditional methods for synthesizing aromatic nitriles mainly involve the diazotization of aniline followed by the Sandmeyer reaction. For less complex benzonitriles, they can be directly oxidized from toluene compounds under the action of NH3. However, these methods have significant limitations: the reaction conditions are harsh, the substrates must be relatively simple with fewer substituents, and toxicity is high. The following introduces commonly used laboratory methods.
7.1a Reaction of Aromatic Halides with Cyanosulfonyl Compounds to Prepare Corresponding Aromatic Nitriles
About 14.7 g (0.05 mol) of 5-bromo-4-chloro-2-methoxybenzoic acid ethyl ester, 5.4 g (0.06 mol) of copper (I) cyanide, and 8 ml of dimethylformamide are heated at 190°C for three hours while stirring under nitrogen atmosphere. After cooling, the reaction mixture is stirred well with 250 ml of methylene chloride and 250 ml of 2N hydrochloric acid. The insoluble portions are filtered off with suction filtration and the layers are separated in a separating funnel. The methylene chloride solution is washed neutral with water and then concentrated by evaporation. The obtained residue was re-crystallized from methylene chloride/hexane to give pure 4-chloro-5-cyano-2-methoxybenzoic acid ethyl ester.
Ref.: Frontpage/Claim: 59938; Patent; CibaGeigy Corporation; Publ.: US4559349 A1 (1985/12/17), Appl.: US1984-586493 (1984/03/05)
7.1b Aromatic Halides with KCN or Zn(CN)2 Under Palladium Catalysis Achieve Cyanide Substitution Reaction
This type of reaction commonly uses catalysts and ligands such as Pd(PPh3)4, Pd(OAc)2/PPh3, Pd2dba3, etc. DMF or NMP are commonly used solvents.
Example 1
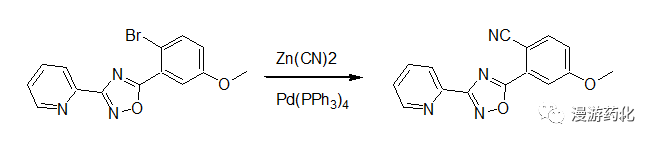
In a similar fashion, a mixture of 3-(2-pyridyl)-5-(2-bromo-5-methoxyphenyl)-1,2,4-oxadiazole (33.2 mg, 0.1 mmol), zinc cyanide (17.6 mg, 0.15 mmol), and Pd(PPh3)4 (11.5 mg, 0.01 mmol) in N,N-dimethylformamide (1 mL) was heated under an argon atmosphere at 80°C for 16 hours. After cooling, the reaction mixture was poured into water, and the crude product was extracted with dichloromethane. Silica gel chromatography using 50 percent ethyl acetate in hexane afforded 3-(2-pyridyl)-5-(2-cyano-5-methoxyphenyl)-1,2,4-oxadiazole.
Ref.: Patent; Wagenen, Bradford Van; Publ.: US2003/55085 A1 (2003/03/20), Appl.: US2002-76618 (2002/02/19)
Example 2
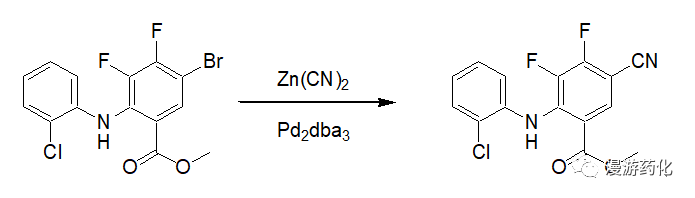
A mixture of 5-bromo-2-(2-chlorophenylamino)-3,4-difluorobenzoic acid methyl ester (14)(3.01 g, 7.99 mmol), 1,1′-bis(diphenylphosphino) ferrocene (dppf) (93 mg, 0.162 mmol), Pd2dba3 (73 mg, 0.080 mmol), and Zn(CN)2 (573 mg, 4.78 mmol) in 1-methyl-2-pyrrolidinone (NMP: 4.5 ml) was heated in a sealed tube reactor. After 20 hours, the reaction mixture was cooled to room temperature, quenched by the addition of 8 ml of a 4:1:4 (volume) mixture of saturated NH4Cl, concentrated NH4OH, and water. The solution was extracted with a mixture of EtOAc/THF. The combined organic extracts were washed with a mixture of 4:1:4 (volume) of saturated NH4Cl, concentrated NH4OH, and water, and then brine. The organic layer was dried (MgSO4) and concentrated. Purification by flash column chromatography using the Biotage system (twice: 100 percent hexanes to 35 percent CH2Cl2 in hexanes, then 30 percent CH2Cl2 in hexanes) provided 1.33 g (52 percent) of the desired product.
Ref: Patent; Wallace, Eli; Publ.: US2005/54701 A1 (2005/03/10), Appl: US2004-929295 (2004/08/30)
7.2 Under Copper Catalysis, Aromatic Halides or (TfO-) and K4[Fe(CN)6] Reaction Achieves Cyanide Substitution
Recently, Thornds Schdreind, Alexander Zapf reported a method for achieving high yields of cyanide compounds under the copper catalysis of aromatic halides or (TfO-) and K4[Fe(CN)6]. The authors conducted a series of experiments using different copper catalysts, as well as different ligands and solvents, to achieve the best experimental conditions, namely: Cu(BF4)2·6H2O as catalyst, DMEDA as ligand, and DMAc as solvent.
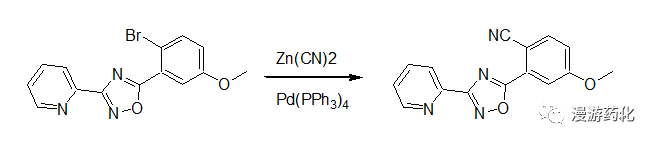
No condition details were available in this literature. The general reaction conditions given by the author were: 2.0 mmol aryl halide, Cu(BF4)2·6H2O (0.1 eq), 20 mol% dry K4[Fe(CN)6], 2 mL DMAc, 20 mol% KI, 20 mol% Na2CO3, 100 mol% DMEDA.
Reference: Tetrahedron Letters. 46 (2005) 2585-2588
7.3 Microwave Reaction of Aromatic Halides Nitration
In the 7.1 reaction example, these direct substitutions mostly use high-temperature reactions; recently, some have developed using microwave reactions for this substitution.
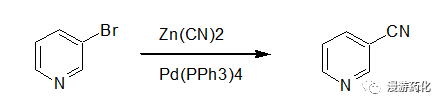
A dried heavy-walled pyrex tube was charged with organo-bromide (0.2 mmol), Zn(CN)2 (23.5 mg, 0.2 mmol), and Pd(PPh3)4 (6.9 mg, 6.0 μmol) in DMF (1 ml). The reaction mixture was flushed with nitrogen, and the screw cap tightened thoroughly before mixing with a Whirlimixer. The reaction mixture was exposed to microwave irradiation (60 W) for 2 min (for 2g 2.5 min). The reaction tube was allowed to reach room temperature before the reaction mixture was diluted in EtOAc (60 mL) and washed with water. The organic phase was dried, and the solvent was removed under reduced pressure. The crude product was purified by column chromatography to give the pure nitrile.
Reference:: J. Org. Chem. 2000, 65, 7984-7989.
8. Oxime Dehydration to Nitriles
Aromatic or aliphatic aldehydes can be converted to the corresponding nitriles by transforming them into oximes and dehydrating.
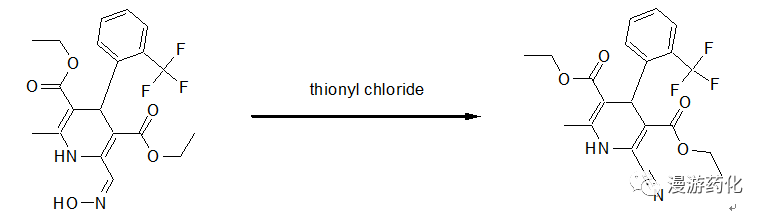
A solution of the powder (0.49 g) of diethyl 2-methyl-4-(2-trifluoromethylphenyl)-6-hydroxyiminomethyl-1,4-dihydropyridine-3,5-dicarboxylate and thionyl chloride (1.5 ml) in dry diethyl ether (1.5 ml) was stirred at room temperature for 30 minutes. After the resultant solution was evaporated to dryness, water was added to the residue, and the mixture was extracted with ethyl acetate. The extract was washed with water, dried over magnesium sulfate, and concentrated under reduced pressure to give brown oil (0.39 g). The oil was purified by column chromatography on silica gel with eluent of 5:1 benzene:ethyl acetate and crystallized with n-hexane to give a yellow powder (50 mg). The powder was further recrystallized from diethyl ether/n-hexane to give crystals of diethyl 2-methyl-4-(2-trifluoromethyl-phenyl)-6-cyano-1,4-dihydropyridine-3,5-dicarboxylate.
Reference: Chem Pharm Bull. 1991, 89-3201.
This content is sourced from the internet, and the copyright belongs to the original author.
