Abstract
Recently, Professor David W. C. MacMillan, a 2021 Nobel Prize winner in Chemistry from Princeton University, reported a novel photonic/nickel cooperative catalysis strategy based on the double decarboxylative cross-coupling (SH2) reaction, which can be used to construct valuable C(sp3)-C(sp3) coupling products from aliphatic carboxylic acids. Detailed mechanistic studies indicate that this reaction involves an SH2 process between Ni(III)-alkyl species and alkyl radicals. Furthermore, this reaction provides a general strategy for efficiently constructing sterically hindered tertiary carbon centers, addressing long-standing challenges in classical coupling reactions.
Main Content
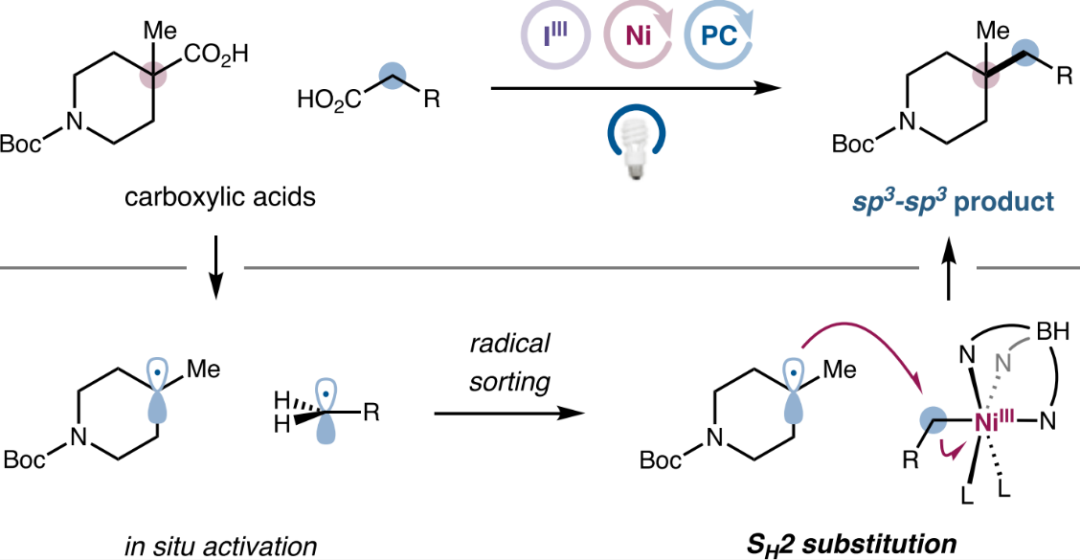
Figure 1. Photonic/Nickel Catalyzed Double Decarboxylative Cross-Coupling (SH2) (Image Source: J. Am. Chem. Soc.)
Transition metal-catalyzed cross-coupling reactions allow the construction of complex compounds through the assembly of simple molecular building blocks. Traditional cross-coupling reactions mainly involve the coupling between classical organic electrophiles and organometallic nucleophiles. In recent years, the emergence of photoredox catalysis has expanded coupling strategies to more substrates containing common natural functional groups, such as halides, alcohols, carboxylic acids, and C-H bonds. Among these coupling building blocks, carboxylic acids have gained widespread attention due to their commercial availability, structural diversity, and stability. Numerous photoredox reactions involving the decarboxylation of aliphatic carboxylic acids have been reported, including alkylation, arylation, amination, and trifluoromethylation. However, achieving high selectivity in cross-decarboxylative C(sp3)-C(sp3) coupling between two different carboxylic acids remains challenging.
Traditional transition metal-catalyzed C(sp3)-C(sp3) coupling is primarily achieved through the formation of bimetallic alkyl complexes. However, the reduction elimination rate of these bimetallic alkyl intermediates is often slow and prone to side reactions such as β-H elimination, carbon-metal bond cleavage, and oxidation of electron-rich alkyls to carbocations. Free radical cross-coupling has emerged as an ideal alternative to traditional metal-catalyzed coupling reactions and has gradually attracted attention in recent years. However, since reactions between free radicals are generally diffusion-controlled, when two radicals have similar lifetimes and are generated at similar rates, a mixture of self-coupling and cross-coupling is obtained (Figure 2A). To address these issues, the authors propose a double decarboxylative cross-coupling strategy (SH2) by selectively combining metals with radicals to generate metal-alkyl species that react with another radical to achieve cross-coupling, where the selective combination of metals and radicals can be controlled by the substitution degree and reactivity of the alkyl radical itself (Figure 2B). Based on this design principle, the authors successfully developed an iron porphyrin-catalyzed cross-coupling reaction between alkyl bromides and redox-active esters, and on this basis, they aim to develop a selective cross-coupling between two carboxylic acid substrates (Figure 2C).
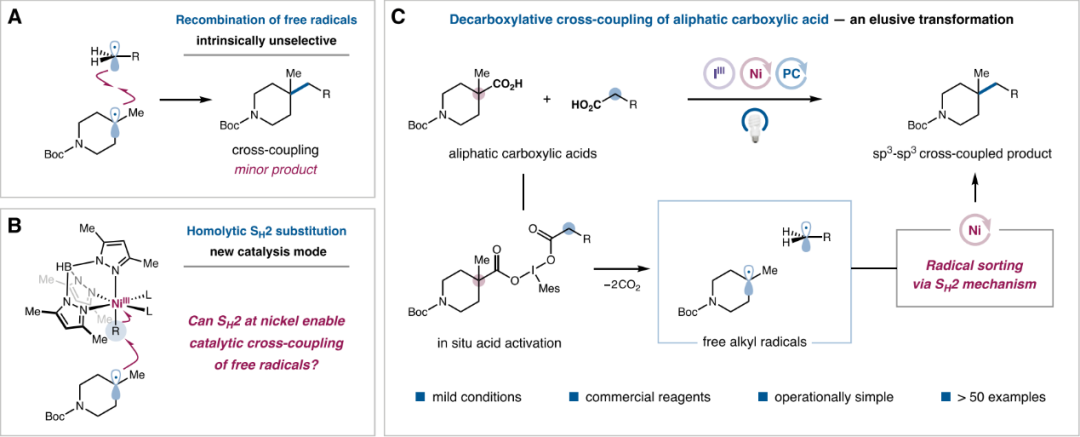
Figure 2. Research Background and Reaction Design Concept (Image Source: J. Am. Chem. Soc.)
First, the authors proposed the design concept and corresponding mechanism for this double decarboxylative cross-coupling reaction (Figure 3B). Carboxylic acid 1 first exchanges with the carboxylic acid ligand on the high-valent iodine 2 to generate the corresponding trivalent iodine species 3. Subsequently, the excited state photosensitizer (4CzIPN or TXO) undergoes energy transfer with the high-valent iodine species, breaking two I-O bonds and undergoing decarboxylation to generate alkyl radicals 4 and 5. At this stage, the Ni(II) complex 6 may selectively capture the less substituted alkyl 4, generating the Ni(III) alkyl complex 7. Finally, the SH2 reaction occurs with radical 5 to yield the cross-coupling product 8 and complete the catalytic cycle.
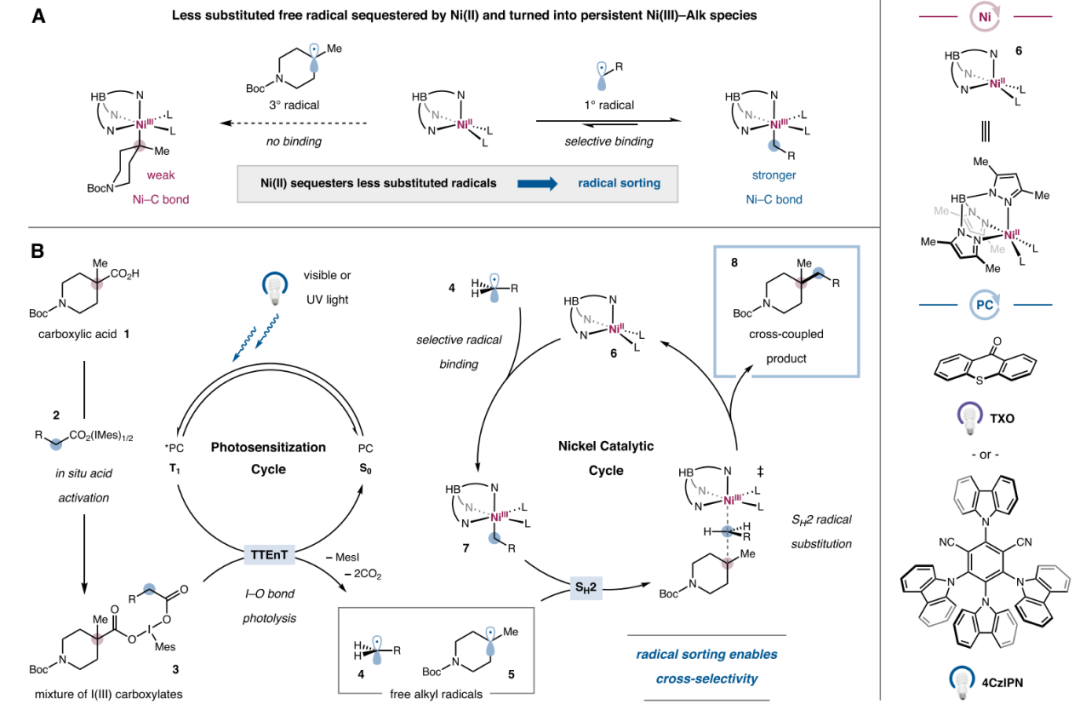
Figure 3. Design of the Double Decarboxylative Cross-Coupling System (Image Source: J. Am. Chem. Soc.)
Subsequently, the authors conducted detailed screening of reaction conditions using N-p-toluenesulfonyl piperidine acid and 2,4,6-trimethyl iodobenzene diacetate MesI(OAc)2 as template substrates (Figure 4). Using Ni(acac)2 as the metal source and the combination of tris(3,5-dimethyl-1-pyrazolyl)borane (K[Tp*]) as the ligand effectively catalyzes the reaction. For photochemical conditions, the authors explored two catalytic schemes, namely, the combination of 365 nm UV light with TXO and 450 nm blue light with 4CzIPN. Compared to the former, visible light conditions are more suitable for some substrates that are unstable under UV light. Finally, by optimizing solvent, temperature, and reaction time, the target decarboxylative coupling product could be obtained with an 85% yield.
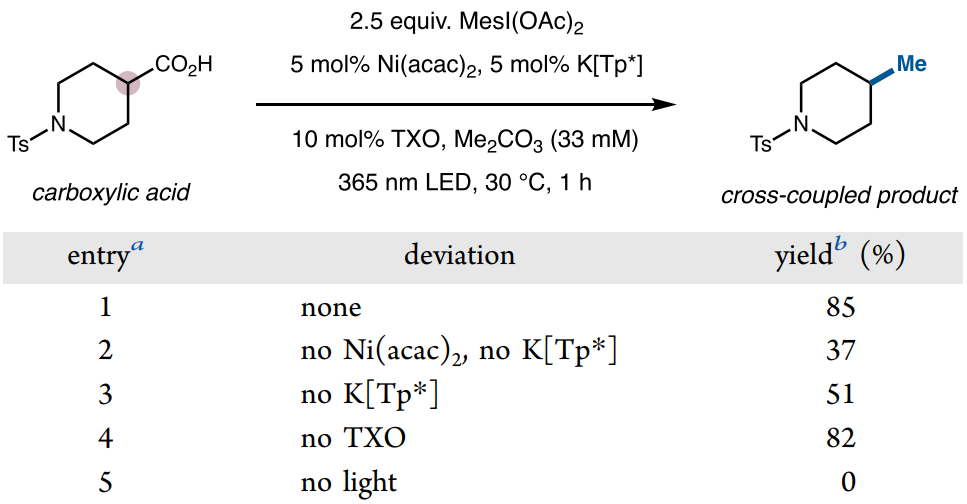
Figure 4. Optimization of Reaction Conditions (Image Source: J. Am. Chem. Soc.)
Under the optimal reaction conditions, the authors examined the substrate scope of the decarboxylative methylation reaction. As shown in Figure 5, this reaction exhibits excellent functional group compatibility. Primary alkyl carboxylic acids containing heterocycles, such as pyridine, benzotriazole, and 1,2,4-oxadiazole, can yield methylation products 9 and 10 with good yields. Numerous secondary alkyl carboxylic acids can smoothly complete this reaction (11–20), where β-lactams and protected and unprotected alcohol hydroxyl groups show good tolerance (11, 12, 18), demonstrating its potential applications in medicinal chemistry. For cyclic carboxylic acids, including nitrogen-containing spiro[3.3]heptane (14, cyclic pentane (15, pyrrolidine (16, piperidine (17–19, and norcocaine (20) exhibit excellent reactivity. For challenging tertiary alkyl carboxylic acids, this catalytic system demonstrates strong applicability, successfully achieving the construction of a series of products containing tertiary carbon centers (21–29). Subsequently, the authors investigated the reactivity of α-amino acids and α-oxocarboxylic acid structural units, which are widely present in biologically active molecules, under standard reaction conditions, yielding decarboxylative methylation products with good yields (30–33), indicating that electron-rich nucleophilic radicals can also be compatible with this system. Finally, the authors examined the applicability of complex drugs and natural products, obtaining methylation products from artemisinin lactate containing peroxide linkages and gibberellin A3 containing two unprotected allyl alcohol groups with moderate to good yields (34, 38). The drug molecule anlisentan containing α-diphenyl fragments was successfully decarboxylatively methylated, indicating that this reaction can be applied to highly sterically congested substrate environments (36).
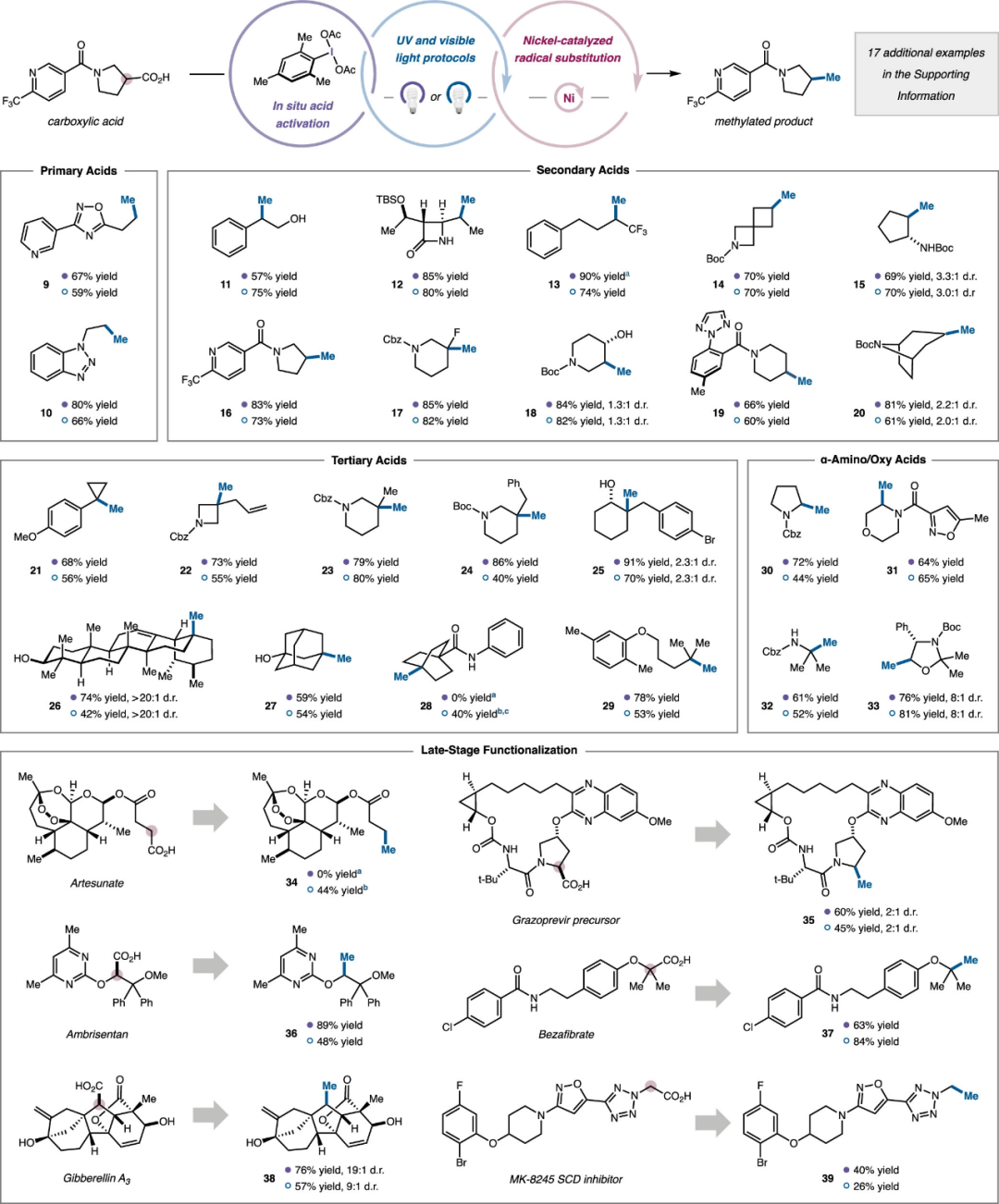
Figure 5. Expansion of Substrates for Decarboxylative Methylation Reaction (Image Source: J. Am. Chem. Soc.)
After completing the decarboxylative methylation reaction using iodobenzoate as the methyl source, the authors examined the reactivity of other dicarboxylic acid iodobenzoates, which can be obtained from MesIO reacting with the corresponding carboxylic acids (Figure 6). The results indicate that isotopically labeled CD3, 13C-methyl can both be applied to this decarboxylative coupling reaction (40, 41). Multi-substituted acetic acid (42–44) and propionic acid (45–46) can complete the corresponding alkylation reactions with moderate to good yields. Finally, the authors successfully constructed a series of tertiary carbon center products using this reaction (47–49).
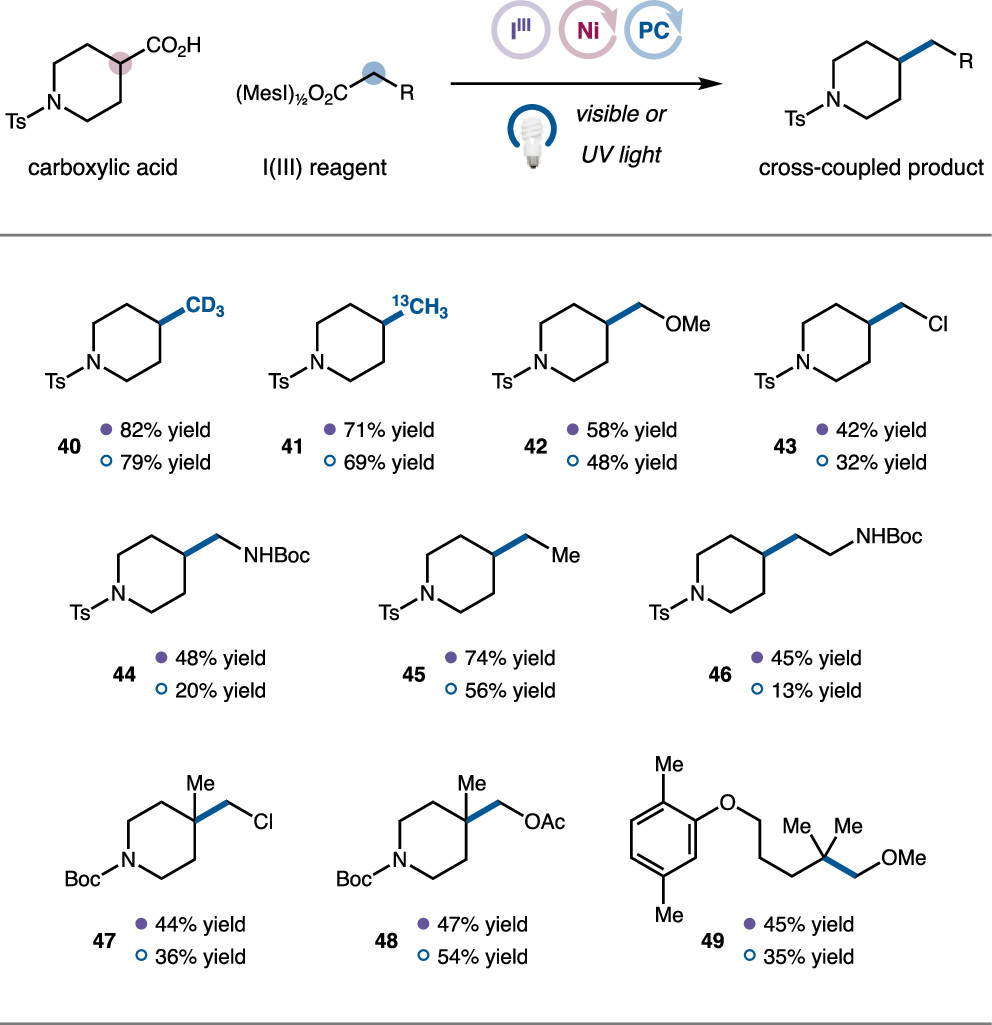
Figure 6. Expansion of Carboxylic Acid Substrates (Image Source: J. Am. Chem. Soc.)
Subsequently, the authors investigated the mechanism of the nickel catalytic cycle in this system. First, kinetic analysis indicates that under UV light conditions, the photosensitization of high-valent iodine species is the rate-determining step of this reaction, and the reaction rate is independent of the concentration of nickel catalysts. Regarding the stereochemical process of this transformation, the authors examined the β-amino acid substrate 50 under various methylation reaction conditions to study the ratio of trans and cis products 15 (Figure 7A). The results indicate that without adding nickel catalysts, a cross-coupling product can be obtained with a yield of 34% and a d.r. value of 1.5:1, likely generated through diffusion-controlled classical free radical coupling. By adding Ni(acac)2 and ligand K[Tp], the yield and d.r. values can be improved to 63% and 2.4:1, respectively. Using the optimal ligand K[Tp*], the final yield can reach 81% with a d.r. value of 3.0:1 for the product. These results indicate that Ni species directly participate in the formation of the C-C bond. Generally, nickel-involved metal-organic reactions typically involve low-valent Ni(0) or Ni(I) species; however, cyclic voltammetry experiments indicate that excess MesI(OAc)2 can effectively oxidize Ni(I) species to Ni(II). To further confirm that active low-valent nickel species are not involved in this catalytic transformation, the authors conducted control experiments by adding aryl halides that easily undergo oxidative addition with low-valent nickel. The results show that the addition of halides does not inhibit this double decarboxylative coupling reaction, and aryl halides can be quantitatively recovered (Figure 7B). The authors further utilized EPR experiments to characterize the existence of Ni species in this system (Figure 7C). In the mixed solution of MesI(OAc)2, Tp*Ni(OAc) complex 55, and TXO, no EPR signal was detected under non-illuminated conditions, indicating that high-valent iodine cannot oxidize Ni(II) complexes under thermochemical conditions. When irradiated with 365 nm UV light, an EPR signal with a g-value of 2.17 appeared, originating from a spin system centered on a transition metal atom. Control experiments indicate that a certain intensity of this signal can also be detected under conditions without TXO, and the signal disappears within seconds after turning off the light source. These results indicate that MesI(OAc)2 generates methyl radicals under 365 nm irradiation, which are captured by Tp*Ni(OAc) to form low-spin d7Ni(III)-Me complexes56a. Finally, the authors conducted similar photonic EPR experiments on a series of iodobenzoates (Figure 7D). Except for MesI(OAc)2, only MesI(O2CEt)2 can produce significant EPR signals (g=2.19), while isopropyl and tert-butyl groups could not yield corresponding Ni-alkyl species, which is consistent with the order of Ni-C bond dissociation energies calculated by DFT.
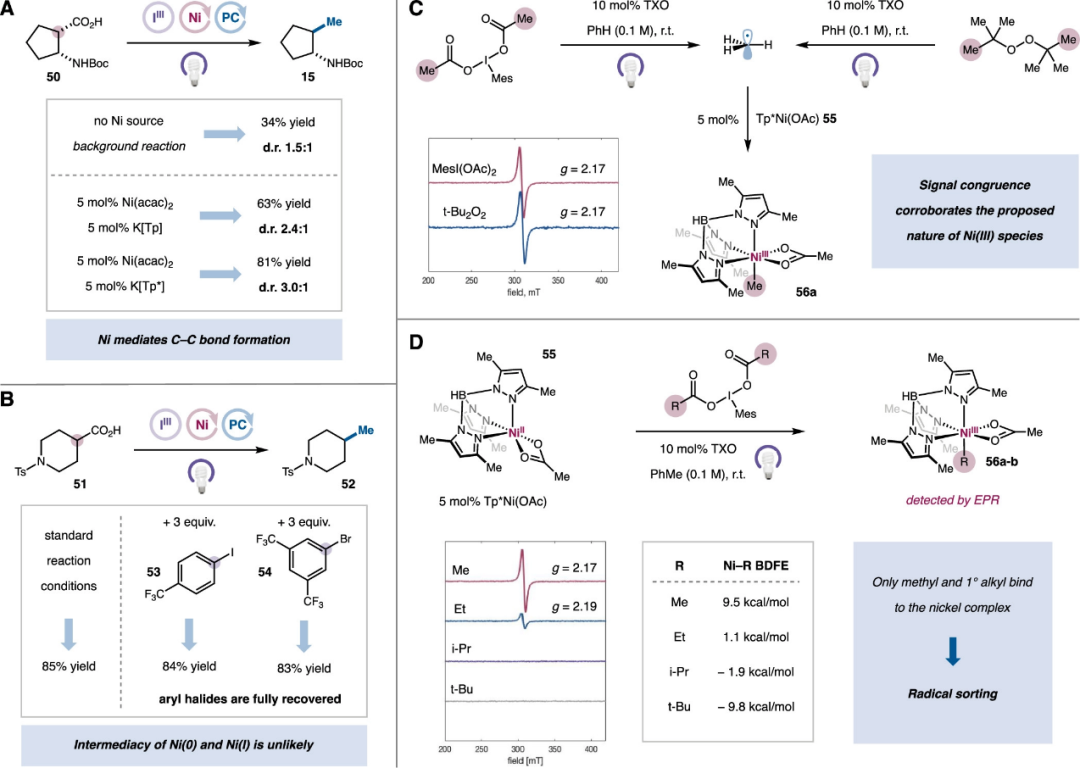
Figure 7. Mechanism Study of the Reaction (Image Source: J. Am. Chem. Soc.)
Conclusion
David W. C. MacMillan‘s research group reported a double decarboxylative cross-coupling reaction of aliphatic carboxylic acids based on the SH2 strategy. In this reaction, alkyl radicals generated by the photosensitization of high-valent iodine are first captured by Ni(II) species, which then undergo SH2 reactions with another radical to achieve selective decarboxylative coupling. This catalytic system exhibits excellent functional group compatibility and can be used to construct tertiary carbon center compounds that are difficult to obtain through traditional metal-organic reactions.
For detailed references:
Artem V. Tsymbal, Lorenzo Delarue Bizzini, David W. C. MacMillan*. Nickel Catalysis via SH2 Homolytic Substitution: The Double Decarboxylative Cross-Coupling of Aliphatic Acids. J. Am. Chem. Soc., 2022. https://pubs.acs.org/doi/10.1021/jacs.2c08989

Long press or scan the QR code on the left to view the original text