Mitochondrial Signal Transduction (Part 3)

Last time: Mitochondrial Signal Transduction (Part 1)
Last time: Mitochondrial Signal Transduction (Part 2)
15
Mitochondrial Communication with Organelles
MIPS has functional interactions with the endoplasmic reticulum, lysosomes, peroxisomes, lipid droplets, and other organelles. Many studies have confirmed this. Mitochondrial metabolism is directly supported by surrounding organelles, which provide various substrates, lipid intermediates, and ionic signals. These ionic signals not only supply substrates but also convey information about the overall state of the cell. In particular, inputs from the cell nucleus provide hundreds of proteins that maintain and endow the mitochondrial molecular sensing mechanisms, fusion/fission dynamics, and motility mechanisms, which influence the tendency of mitochondria to adopt certain network structures.
Mitochondrial cortisol synthesis is a typical example of inter-organelle interdependence, requiring the transfer of cholesterol from lipid droplets to mitochondria, followed by entry through the mitochondrial membrane. Steroid intermediates are then transported from the mitochondria to the endoplasmic reticulum and back to the mitochondrial matrix, ultimately synthesizing cortisol. The synthesis of mitochondrialIMM cardiolipin is similarly dependent, with lipid intermediates shuttling between theERMES andER-mitochondrial complex (EMC) at the mitochondria-associated membranes (MAMs), traversing between mitochondria and the endoplasmic reticulum. Redox-based, timely, localized, and pulsed communication between mitochondria and the endoplasmic reticulum can also propagate signals from individual mitochondria to the endoplasmic reticulum and other mitochondria. These examples illustrate the functional interdependence of mitochondria and other organelles, as well as the existence of conserved mechanisms for information exchange, transmittingMIPS states to other organelles and vice versa.
16
Section: Mitochondrial Signal Integration
Having described the molecular mechanisms that allow mitochondria to perceive and dynamically respond to intracellular and systemic inputs, we discuss here the mechanisms that enable mitochondria to communicate and exchange information with each other and with other organelles. WhenMIPS interacts physically and functionally as a whole with other organelles, it forms a biochemical and energetic condition distribution characterization of the cell and organism. In turn, the ability to sense and integrate this information is adaptive, allowing mitochondria to adjust and optimize their morphological and functional states to alter intracellular and environmental conditions.
It is worth noting that soluble communication mechanisms undoubtedly complement more complete forms of mitochondrial communication, including membrane fusion and more or less complete merging of mitochondria. If diffusion signals and transient protein exchanges are akin to “kiss-and-run”, then more stable physical contacts between mitochondria, such asIMJs and nano-tunnels, may reflect “engage-and-hold,” while complete mitochondrial fusion resembles “marry-and-mix.” Therefore, mitochondrial interactions may be relatively transient (with ionic outflow lasting a few milliseconds), selective (with nano-channels connecting only to one receptormito), and reversible (inter-organelle chains can dissociate). The nature of these interactions is consistent with other plasticity mechanisms in biology, such as those regulating synaptic function in neural networks, which similarly integrate inputs and compute information.
However, the ultimate units of evolution and adaptation are not mitochondrial networks or individual cells, but the groups of cells that constitute the organism. Therefore, the goal of mitochondrial sensing and integrating information must be to optimize the organism’s self-adaptiveness and health. Biologically, ifMIPS perceives and integrates information that is subsequently transmitted to other parts of the cell and organism, this becomes possible. This logic leads us to consider how mitochondrial signals convert and transmit mitochondrial inputs into meaningful cellular and organismal output signals (MIPS Step 3/3)..
17
Mitochondrial Signals
Some mature and emerging signaling pathways link mitochondrial behavior to gene expression within the nucleus. Furthermore, outside the cell,MIPS releases signals into the systemic circulation, influencing metabolic processes in adjacent cells and distant target organs. Several elements of mitochondrial signaling have been extensively reported elsewhere, such as apoptotic signaling,ROS-mediated signaling, and metabolic intermediates. Here, we provide only a brief overview of these areas and extend the discussion of mitochondrial signaling to a broader mitochondrial output as cellular and/or systemic outputs, including small metabolites, proteins,DNA, steroid hormones, and non-molecular signals (including heat) (Figure 6). We also discuss how the potency and specificity of signaling are influenced by the subcellular localization of signaling mitochondria.
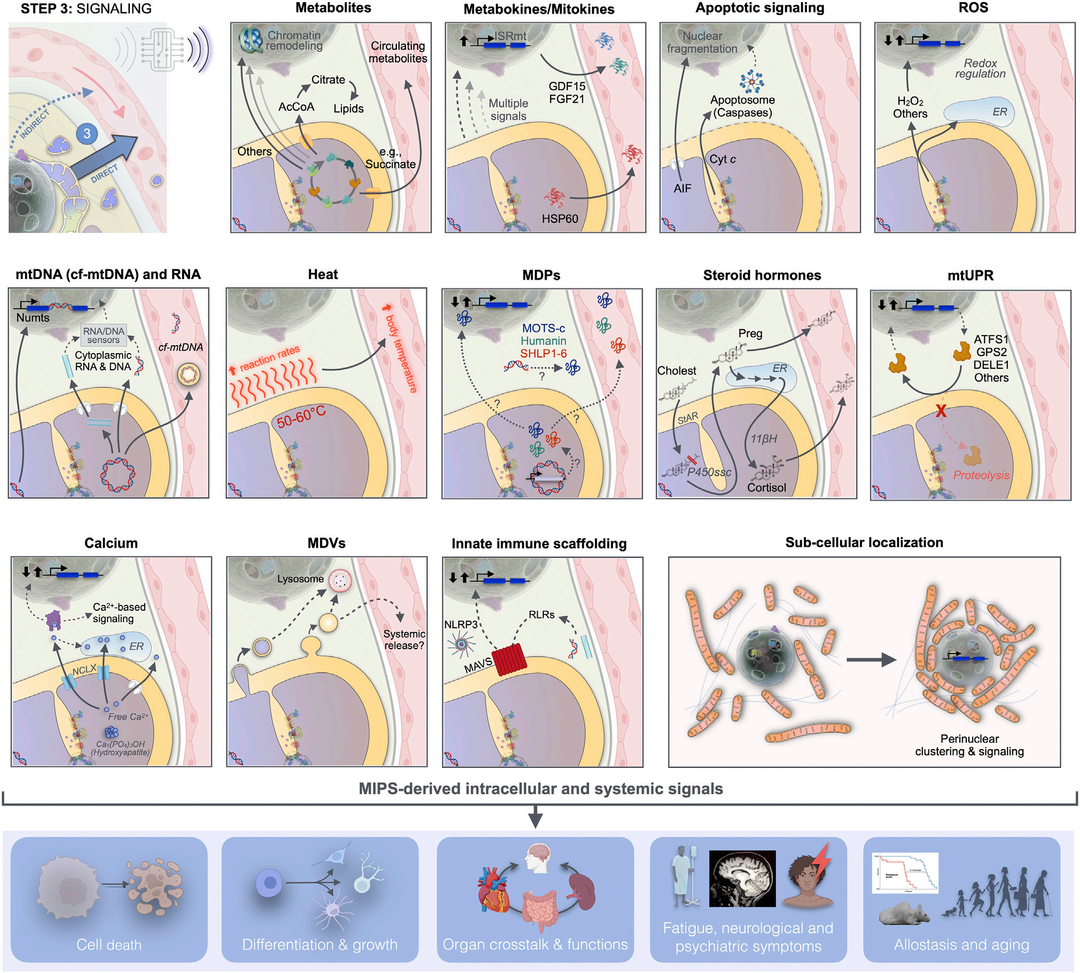
Figure 6 MIPS Step 3: Signals
Mitochondrial synthesis and release affect signals that influence cellular and organismal functions. Mitochondrial signals arise from different mitochondrial compartments, reaching the cytoplasm, nucleus, and other organelles, where they induce autonomous cellular responses. These responses are either directly transmitted to the systemic circulation as mitochondrial-derived metabolites and fission factors or indirectly through transcriptional regulation of nuclear genes encoding metabolic factors or other hormone-like mediators to the systemic circulation.mitto -nuclear signaling is a form of signal amplification and integration.MIPS transforms metabolic signals into extracellular protein secretory factors, enabling mitochondria to signal their state beyond the confines of the cell they reside in.
lAcCoA: Acetyl-CoA; AIF: Apoptosis Inducing Factor; ATFS1: Activating Transcription Factor associated with Stress-1; cf-mtDNA: Cell-free mitochondrialDNA; Cholest: Cholesterol;Cyt c: Cytochromec; DELE1: DAP3 Binding Cell Death Enhancer1; ER: Endoplasmic Reticulum; FGF21: Fibroblast Growth Factor21; GDF15: Growth Differentiation Factor15; GPS2:G Protein Pathway Inhibitor2; HSP60: Heat Shock Protein60; ISRmt: Integrated Stress Response;MAVS: Mitochondrial Antiviral Signaling;MDVs: Mitochondria-Derived Vesicles;MPDs: Mitochondrial-Derived Peptides;NLRP3:NOD-like receptor thermal protein domain associated protein3; Numts: Nuclear mitochondrialDNA fragments;Preg: Pregnenolone;; P450ssc: Side chain cleavage enzyme cytochromeP450; RLRs, RIG-l-like receptors;StAR:Steroid Acute Regulatory Protein;UPRmito: Mitochondrial Unfolded Protein Response;11ßH, 11β-hydroxylase (Mitochondrial CytochromeP450 11B1).
18
Apoptotic Signals
The term “mitochondrial signal” was first used in1999 in connection with the release of the pro-apoptotic mitochondrial outputCyt c. Cyt c is a small heme protein that is typically found in theIMS, transferring electrons betweenOxPhos complexesIII andIV. However, in response to specific input signals such asROS, high[Ca2+] and low[ATP], particularly in fragmented and poorly connected mitochondrial networks, mitochondria undergo permeability transition by opening thePTP. Opening of thePTP triggers the cytosolic release ofCyt c, where it interacts with pro-caspases and activates, along with other mediators of the intrinsic apoptotic pathway, including apoptosis-inducing factor (AIF) and endonucleaseEndoG, which translocate to the nucleus, nuclear genomic fragments, andSmac/Diablo. In cancer cells, the release ofCyt c during non-lethal permeability transition (i.e.,“blip mode“) can also play a non-apoptotic signaling role, including activating nuclearATF4-dependent integrated stress responses (ISRs; see below). To prevent the assembly of pro-apoptotic complexes at theOMM, mitochondria can also recruit anti-apoptotic proteins from theBcl2 family. Functionally, the opening ofPTP is closely related to the release and signaling of mitochondrialCa2+, which is increasingly under the control of emerging ridge-regulating mechanisms. Therefore,MIPS encompasses many powerful cellularlife-or-death signals that are coordinated for release based on an integrated characterization of mitochondria and cytoplasm.
19
Mitochondrial Metabolic Signals
The epigenome is the language of mitochondria. It is likely that the endosymbiotic origin of mitochondria andMIPS preceded the development of histone codes, thus the current epigenetic nuclear mechanisms have evolved to integrate gene expression with the metabolic state of the cell in the context of mitto-nuclear communication. Consequently, the nuclear genome is densely wrapped in rich histones (mainlyH2A, H2B, H3 andH4), which have hydrophilic tails that are extensively post-translationally modified by the metabolic chemical modifications of the nuclear periphery and nuclear environment.
Histone-modifying enzymes alter most substrates or cofactors required for histone structure and downstream gene expression, which are direct products of mitochondrial metabolism. These include metabolites of theTCA cycle and one-carbon metabolism. For instance, histone methyltransferases (HMTs) andDNA methyltransferases (DMTs) methylate histones andDNA, respectively, requiring S-adenosylmethionine (SAM), produced from serine metabolism as part of the folate cycle and one-carbon metabolic pathway. Conversely, reverse demethylation reactions require cofactorsα-ketoglutarate (αKG), aTCA cycle metabolite. Some mitochondrial-derived metabolites participate in post-translational modifications (PTMs) of histones (and other proteins). These include lactate (i.e., lactylation), a metabolite produced from glycolysis, whose concentration increases when mitochondrialOxPhos is impaired; dopamine (i.e., dopaminylation), which is metabolized by monoamine oxidase associated withOMM, which also involves the respiratory chain;β-hydroxybutyrate (i.e., β-hydroxybutyrylation), synthesized in the mitochondrial matrix under low carbohydrate conditions, and other metabolic processes.
Mitochondrial metabolites areMIPS outputs of epigenomic modifications. The Rho0 cells with mtDNA deletions were initially used to demonstrate that mitochondrial outputs can alter nuclearDNA methylation. In a similar model, a series of pathogenic human cell lines with different mutation loads (i.e., heterogeneity) were compared, ranging from0% to100% heterogeneity, to dose-dependently influenceDMT gene expression and global transcriptional characteristics. In the same model, mtDNA heterogeneity altered levels of acetyl-CoA andaKG and led to downstream changes in H4K16ac and H3K9me3 states. Acute mtDNA depletion in immortalized cells also triggered a physiologically significant reduction in mitochondrial acetyl-CoA and downstream histone acetylation, illustrating the scope ofMIPS epigenomic effects. Finally, a longitudinal study tracking DNA methylation changes over several months in primary human fibroblasts indicated that genetic and pharmacologicalOxPhos defects led to conserved, age-related high methylation and low methylation at thousands of genomic loci encoding developmental programs and cell-cell signaling components. An openly available multi-omics longitudinal dataset can be used to explore the effects of bioenergetic disturbances on the epigenome and transcriptome of aging human fibroblasts. In summary, these findings elucidate some mechanisms by which intrinsicmtDNA related andOxPhos inputs are transduced into epigenomic remodeling outputs.
However, how MIPS-induced molecular and epigenetic modifications maintain precise targeting in time, space, and molecular terms, as well as their functional consequences on gene expression and cellular phenotypes, remains unclear. Additionally, the presence of various activeTCA cycle enzymes directly in the nucleus complicates this scientific challenge. The presence of mitochondrial enzymes in the nucleus, primarily recorded in cancer cell lines to date, suggests that at least in some cell types, mitochondria may not be the only source of chromatin-modifying metabolites.
In recent years, other mitochondrial metabolites and molecular features have emerged as widespread intracellular signals. For example,TCA cycle metabolite succinate and fumarate levels are regulated through theOxPhos system via electron transport, fundamentally regulated by succinate hydratase and succinate dehydrogenase (Martinez-Reyes andChandel have conducted comprehensive reviews). These metabolites are released into the cytoplasm, and their regulation involves signaling pathways for hypoxia sensing, immune activation, inflammation, and oncogenic transformation.TCA cycle metabolites are also converted by enzymes into metabolic derivatives such as methylene succinate and 2-hydroxyglutarate. Methylene succinate is produced from the TCA cycle metabolite aconitate through aconitate decarboxylase and then acts on mitochondrial enzymes, such as inhibiting succinate dehydrogenase, or on transcription factors in the cytoplasm/nucleus, such as inhibitingNF-kB signaling.2-hydroxyglutarate (2-HG) has two isomers produced byαKG through mitochondrial or cytoplasmic malate dehydrogenase (respectively,MDH2 andMDH1) in an NADH-dependent manner and is promoted under acidic pH conditions. In the nucleus,2-HG subsequently inhibits histone tail andDNA demethylation through t10-11 hydroxylases (TET1-3), playing a crucial role in influencing tumorigenesis and fate transitions in immune activation. Besides soluble metabolites, larger mitochondrial lipids also play significant signaling roles. For instance,IMM lipid cardiolipin participates in various cellular signaling events, translocating toOMM during stress, and serves as a signaling platform associated with mitochondrial autophagy, apoptotic signaling, and other functions.
In addition toTCA cycle flux, NADH/NAD+ ratios, andpH, the presence of carrier proteins onIMM can also affect MIPS metabolite signaling. For instance, in mesenchymal stem cells, age-related changes in citrate carrier expression regulated the cytoplasmic output of acetyl-CoA, driving histone acetylation levels, increasing chromatin accessibility, and affecting stem cell differentiation. Therefore, the nature and intensity of mitochondrial output may be regulated not only by rapid changes in the flux of specific mitochondrial metabolic pathways but also by the relatively stable composition and abundance ofIMM carriers and transporters (although stability is susceptible to change).
Extracellularly, metabolites also act in a non-autonomous manner. A well-studied example is succinate, a mandatory intermediate of the mitochondrialTCA cycle, influenced by oxygen tension, DpH+DJm, and ATP demand, accumulating according to the redox state balance of coenzymeQ. It has been reported that succinate emits extracellular and systemic signals through at least one cell surfaceGPCR, namely succinate receptor1 (SUCNR1), on immune cells and other cell types to regulate inflammation processes. In target immune (and possibly other) cell types, succinate can also enter through monocarboxylate transporter1 (MCT1), inhibiting TCA cycle activity and signaling pathways, thus suppressing interferon secretion. Therefore, the export of metabolites from MIPS has extensive autonomous and non-autonomous effects on the epigenome, nuclear gene expression, and cellular behavior.
Another mitochondrial metabolite is well known in circadian biology: melatonin (n -acetyl-t-methoxytryptamine). Melatonin is evolutionarily an ancient bacterial molecule with strong antioxidant properties predating endosymbiosis. Mitochondria not only containMT1 melatoninGPCR, but also catalyze two enzymatic reactions through mitochondrial matrix enzymes (arylalkylamine N -acetyltransferase [AANAT] and acetylserotonin methyltransferase [ASMT]) to synthesize melatonin from L-tryptophan (with 5-hydroxytryptamine as an intermediate). Like other mitochondrial metabolites, systemic melatonin levels exhibit strong diurnal variations (undetectable during the day, peaking at night). It can regulate the sleep/wake cycles of some animals, and oral administration in humans may potentially regulate sleep onset. Thus, mitochondrial-derived melatonin can act locally in an autocrine manner and in a paracrine manner between cells/neuron, as well as through release into the bloodstream, demonstrating the broad influence of MIPS-derived metabolites/hormones in mammalian physiology. In summary, mitochondrial-derived metabolic outputs represent complementary signals that integrate the bioenergetic state of MIPS into understandable signals of core cellular signaling mechanisms that coordinate a wide range of cellular and organismal behaviors.
20
Mitochondrial ROS Signals
ROS is a diffusible molecule, particularly hydrogen peroxide (H2O2), produced by the breakdown of superoxide anions (O2.-) from the antioxidant systems in the matrix andIMS. MitochondrialROS primarily originates fromOxPhos complexesI andIII, entering the cytoplasm and nucleus, where they trigger redox-sensitive gene regulatory processes. MitochondrialROS signaling and its measurement guidelines have been detailed previously, so here we primarily focus on the latest advances in this field.
Mitochondrial reactive oxygen species regulate various states and systemic signals within mitochondria. For example, in brown adipose tissue, the production of mitochondrialROS modifies the translation ofCys253 ofUCP1, enhancing uncoupling and achieving thermoregulation, while pharmacologically reducing mitochondrialROS generation with MitoQ can preventIMM uncoupling and thermogenesis. In adipocyte heme synthesis in the mitochondrial matrix,H2O2 oxidizes bilirubin to form biliverdin, which is transported out of mitochondria by ATP-binding cassette (ABC) transporterABCB10. In secretory pancreaticβ cells, glucose-stimulated insulin secretion is similarly driven by the accumulation ofH2O2, indicating that mitochondrialROS signals within mitochondria and cells trigger the release of systemic endocrine signals such as insulin.
Perhaps due to the central role of oxygen in the evolution of aerobic organisms, mitochondrial-derivedROS has widespread impacts on nuclear transcriptional regulation. In cultured cells, increased production ofROS can lead to respiratory chain dysfunction, which can also be simulated by adding mitochondrial-targeted redox cycling agents like paraquat (MitoPQ), sufficient to activate mitogen-activated protein kinases (MAPK) proteins, includingJNK signaling, inducing secondary signals such as the release of nuclear chromatin into the cytoplasm. Similarly, inducing high levels of temporally controlledROS in mitochondria using chemogenetic tools can elevate nuclear hydrogen peroxide levels and induce telomere damage. In mice, silencing mitochondrial matrix antioxidant enzyme manganese superoxide dismutase (MnSOD) during development indicates that mitochondrial-derivedROS activates cytoplasmic/nuclearNrf2 andPPARγ/PGC-1α pathways, leading to sustained adaptive responses in adult animals. During embryonic and postnatal development, treatment with low-dose serotonin ketone (a complex I inhibitor that increases mitochondrialROS release) in mice also yielded similar results, altering nuclearDNA methylation and coat color. In aging human fibroblasts, mitochondrial signals transmitted throughROS are necessary and sufficient for activatingNf-kB pathways and aging characteristics (including the senescence-associated secretory profile[senescence-associated secretory profile (SASP)]). In fact, treatment of human fibroblasts with Parkin-overexpressing/FCCP-treated mitochondria prevented the occurrence of aging characteristics, providing compelling evidence thatMIPS signals (including but not limited toROS) are necessary to trigger complex cellular states such as aging. Furthermore, the SASP can spread the aging phenotype to neighboring bystander cells both in vitro and in vivo, illustrating that mitochondrial signals are one of many pathways through which non-autonomously propagate systemically to influence organism behavior and lifespan.
In addition to mitochondrial-nuclear signaling, ROS produced by individual mitochondria also contribute to communication with the endoplasmic reticulum. Even in distant neuronal branches far from the cell body, mitochondrialROS also contribute to local synaptic activity. In response to photodamage of the plasma membrane, mitochondria at the damage site also respond in adrp1-dependent manner, increasing the production ofROS that promote repair. Thus, the site-specific action of mitochondrialROS at subcellular locations highlights the importance and potential specificity of localMIPS ROS outputs as drivers of gene regulation and cellular function.
21
Mitochondrial Synthetic Hormones and Stress-Related Hormones
One of the most powerful types of hormones in mammals is steroid molecules, broadly classified into three main categories: (1) Gonadal hormones that determine sex, including testosterone, estrogen, and progesterone; (2) Stress hormones produced by the adrenal gland that promote stress adaptation, including glucocorticoids and mineralocorticoids; (3) Neurosteroids produced by the nervous system. Their release is regulated by trophic pituitary hormones from the brain.Adrenocorticotropic hormone, ACTH, follicle-stimulating hormone, FSH; In hormone-producing cells, luteinizing hormone (LH) is mediated by GPCR-coupled cyclic AMP protein kinase A (cAMPPKA) orCa2+-KC pathways. In steroidogenic tissues, the rate-limiting step for synthesizing all steroid hormones occurs within mitochondria.
Mitochondria produce steroid hormones from cholesterol, which is the initial substrate for all steroids. The entry of cholesterol through theOMM andIMM requires microtubule and microfilament dynamics as well as protein synthesis, completed by the steroid acute regulatory protein (StAR, derived from theSTARD1 gene) in theOMM. Mitochondria inputStAR through theTIM/TOMM transport complex, leading to its proteolytic degradation, whileStAR stabilizes in theOMM, associating withTom2 andVDAC2, transferring cholesterol to the matrix-facingIMM side chain cleavage enzyme cytochromeP450 (CYP450scc). ThenCYP450scc catalyzes the rate-limiting reaction of steroidogenesis, converting cholesterol into progesterone, which is a common precursor for all steroid hormones. Pregnenolone synthesized in the matrix is subsequently output to the endoplasmic reticulum, where other enzymes sequentially catalyze its conversion into progesterone and other steroid intermediates. In the steroidogenic mitochondria of the adrenal cortex, downstream steroid intermediates may return to the mitochondrial matrix viaMAM, where terminal reactions are catalyzed by the mitochondrial matrix enzyme 11b-hydroxylase (11bH, also known as“mitochondrial cytochromeP450 11B1, encoded byCYP11B1 in humans) to produce cortisol. Mitochondrial synthesis of systemic steroids occurs rapidly within minutes, and this synthesis halts just as swiftly. This process occurs rapidly, is redox-sensitive, and is protein input-dependent, illustrating the influence of various intrinsic factors on mitochondrial steroidogenesis output. The evolutionary basis for the mitochondrial localization of steroid formation remains uncertain but may involve unique reductive conditions in the mitochondrial matrix.
22
Intracellular Mitochondrial Genome Signals
CircularmtDNA typically exists in the mitochondrial matrix, isolated from the cytoplasm by two layers of mitochondrial membranes. However, the enclosed spaces within the mitochondrialIMM andOMM can naturally be disrupted under certain physiological conditions. This includes instability ofmtDNA triggered by partial loss of the mitochondrial-associated proteinTFAM, which activates the expression programs of mtDNA-dependent antiviral genes in the nucleus.mtDNA release is a relevant signaling mechanism, as both immune and non-immune cells’ cytoplasm and extracellular surfaces containDNA sensors that can recognizemtDNA fragments as damage-associated molecular patterns (DAMP). DNA (viral, bacterial, andmtDNA) is sensed by various innate immune receptors, includingcGAS (cyclicGMP-AMPsynthase),TLR9 (Toll-like receptor9),NLRP3 (NOD-, LRR-, and pyrin domain-containing protein3),andAIM2 (absent in melanoma). The sensing of mtDNA triggers a signaling cascade that converges on the transcription factors that produce cytokines and interferons, includingIRF3/7,MAPK, andNFkB, or participates in the processing and secretion ofCaspase-1 to releaseIL-1β andIL-18. Mitochondrial double-strandedRNA (mt-dsRNA) cytoplasmic release can also act as aDAMP, detected byRIG-I-like (RLR) receptorsRIG-I andMDA5. Once involved, these sensors translocate to mitochondria and activate mitochondrial antiviral signaling proteins (MAVS), which assemble into filaments on the mitochondrial surface in a membrane potential-dependent manner, acting as an antiviral signaling platform.
Current research on innate immune signaling suggests that the mitochondrial network is both a source of stimulating ligands and a major signaling hub for four main families of pattern recognition receptors (TLRs,NOD,RLRs, and cytosolic DNA sensors [CDSs]). Most actions of exogenous (bacterial, viral) nucleic acids and endogenousmtDNA/mtRNA signaling may be mediated through these pathways. As an example of mitochondrial signaling, when human fibroblasts and cancer cells detect the genotoxic drug doxorubicin,mtDNA damage (sensing) ultimately leads to the cytoplasmic release ofmtDNA fragments (signaling), triggering nuclear DNA repair mechanisms and the activation of cGAS-STING-dependent interferon-stimulated gene (ISG) expression.
Regarding the mechanisms promoting the squeezing ofmtDNA into the cytoplasm, two molecular pathways have been described. One mechanism tested in mouse embryonic fibroblasts and lupus-like disease mice involves the oligomerization ofVDAC in theOMM, forming large pores that allow shortmtDNA fragments of 100 to 400 bp to be ejected into the cytoplasm. Similarly, in bone marrow-derived macrophages, 500-650 bp longmtDNA fragments are cut out from the circular genome by the mitochondrial protein flap-structure-specific endonuclease1 (FEN1) and released into the cytoplasm in a VDAC-dependent manner, activating inflammasomes and cGAS-STING signaling. Another described mechanism for cytoplasmic squeezing ofmtDNA involves pore formation mediated byBAX/BAK in theOMM, leading to protrusions on the mitochondrial surface under genotoxic stress conditions, which appear to also release ds-mtRNAs to activate ISGs in the nucleus. A third non-specific mechanism may involve mitochondrial membrane rupture, often secondary to swelling, as may occur in skeletal muscle of primary mtDNA mutation patients.
A more persistent way for the mitochondrial genome to carry information to the nucleus is through the translocation ofmtDNA fragments into the nucleus, subsequently inserted as nuclearmtDNA inserts (NUMTs, pronounced “nu-mites”) into coding sequences. This process is referred to as “numtogenesis,” traditionally understood as horizontal gene transfer, occurring multiple times throughout the evolution of unicellular and multicellular organisms. Thus, multiple germlineNUMTs are shared among individuals. Within mitochondria,mtDNA gene transfer may also explain how most genes of the ancestral proteobacteria genome migrated to the nucleus, such that 98% of the mitochondrial proteome is now encoded by the nuclear genome.
However, the transfer ofmtDNA sequences to the nucleus may also occur throughout the cell’s life cycle. In yeast, mitochondria lacking the mitochondrial proteaseYme1 (yeast mtDNA escape protein1, a member of the AAA ATPase family) degradeIMS/IMM proteins to regulate mitochondrial ridge dynamics, generating 77-fold more mitochondrial nuclear transferNUMTs, while shortening lifespan by 50%. In human cancer cells, the absence of the human homologYME1L1 results in the production of four times moreNUMTs, recreating the abnormally high number ofNUMTs observed in ovarian tumors and other cancer types. In vitro, stable generation of NUMTs may also occur in primary human fibroblasts at a sustained rate for days to weeks, persisting throughout a human’s lifetime in brain tissue (unpublished data). Whether NUMTs affect nuclear genomic instability and cellular aging is a true, regulated form ofMIPS signaling remains to be determined.
To be continued in: Mitochondrial Signal Transduction (Part 4)
Recommended Reading
Mitochondrial Signal Transduction (Part 1)
Mitochondrial Signal Transduction (Part 2)
The Role of Mitochondria in Immune Responses to Critical Illness
Mitochondrial PO2 Monitoring: Just Around the Corner


Editor: Wu Chunlan
Reviewer: Fu Xiaoyun
Contact Information
24-hour Hotline:
0851-28608514
0851-28609875
Address:
149 Dalian Road, Huichuan District, Zunyi City, Guizhou Province
(Department of Critical Care Medicine, Zunyi Medical University Affiliated Hospital)

Long press to scan the QR code
Follow the public account
Get more exciting content