
▼ Click to Follow Our Official Account

NXP2 Antibody Positive Juvenile Dermatomyositis
Seven-year-old Xiao Yu (a pseudonym) started to show signs of not wanting to walk, unwilling to dress himself, and not wanting to wash his face or comb his hair around February 12 this year, constantly complaining of tiredness, leg pain, and lack of strength. His parents only felt that the child had become very lazy. Recently, Xiao Yu has become increasingly lazy, refusing to continue activities after walking for just one minute, and began to show swelling in both lower limbs, accompanied by significant pain and fatigue. Dark red rashes also started to appear on his body, along with swelling of the eyelids. With questions in mind, Xiao Yu’s mother took him to the Rheumatology Department at Fudan University Children’s Hospital. After detailed questioning, a systematic physical examination, and relevant tests, Xiao Yu was finally diagnosed with NXP2 antibody positive juvenile dermatomyositis. After receiving timely and correct treatment, Xiao Yu regained his vitality. However, his mother still had many questions: what exactly is NXP2 antibody positive juvenile dermatomyositis?
JDM
Juvenile Dermatomyositis

First, let’s understand what juvenile dermatomyositis (JDM) is.
Juvenile dermatomyositis (JDM) is the most common idiopathic inflammatory myopathy (IIM) in children, primarily characterized by typical skin rashes (including heliotrope rash, Gottron’s papules, Gottron’s sign, see Figures 6-7), symmetrical proximal muscle weakness or lack of strength, and elevated muscle enzymes. In addition to these characteristics, children often experience non-specific symptoms such as low-grade fever, malaise, weight loss, and loss of appetite. The exact cause of JDM is unclear but may be related to environmental triggers, immune dysfunction in genetically susceptible individuals, and interactions with specific tissues. The disease primarily affects the skin and muscles but can also involve other organs such as the lungs, joints, heart, gastrointestinal tract, and nervous system, presenting symptoms related to these systems. The common pathological change in all affected tissues is vascular lesions. Treatment often involves a combination therapy regimen, including corticosteroids, immunosuppressants, biologics, or small molecule targeted drugs such as JAK inhibitors. If the acute phase is poorly controlled, it can lead to long-term sequelae such as muscle atrophy, tendon contractures, and calcification, and even pose a life-threatening risk. Currently, the pediatric clinical work still frequently uses the JDM classification scheme and diagnostic criteria established by Bohan and Peter in 1975, which are simple and practical.
JDM Classification Scheme and Diagnostic Criteria
The JDM classification scheme and diagnostic criteria established by Bohan and Peter in 1975 are as follows:
(1) Typical skin changes, including purplish-red skin on the eyelids accompanied by periorbital edema (heliotrope rash) and red scaly rashes on the dorsal sides of the metacarpophalangeal joints and proximal interphalangeal joints (Gottron’s sign);
(2) Symmetrical proximal muscle weakness, possibly accompanied by dysphagia and respiratory muscle weakness;
(3) Laboratory tests: elevated serum skeletal muscle enzyme activity, especially CK and AST;
(4) Abnormal electromyography: potentials, short-term multi-phase waves; fibrillation potentials, positive sharp waves, prolonged insertion potentials; high-amplitude abnormal discharges at rest, etc.;
(5) Abnormal muscle biopsy: muscle fiber degeneration, necrosis, cell phagocytosis, regeneration, eosinophilia, enlarged nuclear membranes, significant nuclear membranes, atrophy of perimysial structures, variable fiber sizes, accompanied by inflammatory exudate.
If items (1) and three or more of items (2) to (5) are present, a diagnosis of JDM can be made. If item (1) is absent but three or more of items (2) to (5) are present, the diagnosis is polymyositis.
In addition to the classic diagnostic criteria mentioned above, the European League Against Rheumatism/American College of Rheumatology (EULAR/ACR) also established classification criteria for adult and juvenile idiopathic inflammatory myopathies (IIM) in 2017. This standard assigns different weightings to rashes, muscle weakness, other clinical manifestations, and laboratory tests based on whether a muscle biopsy is performed (Figure 1). The criteria for diagnosing IIM (probability ≥90%) are: for those without muscle biopsy, a score ≥7.5; for those with muscle biopsy, a score ≥8.7. If the classification diagnostic criteria are met, and the age of onset is <18 years with typical rash manifestations, the diagnosis is JDM.
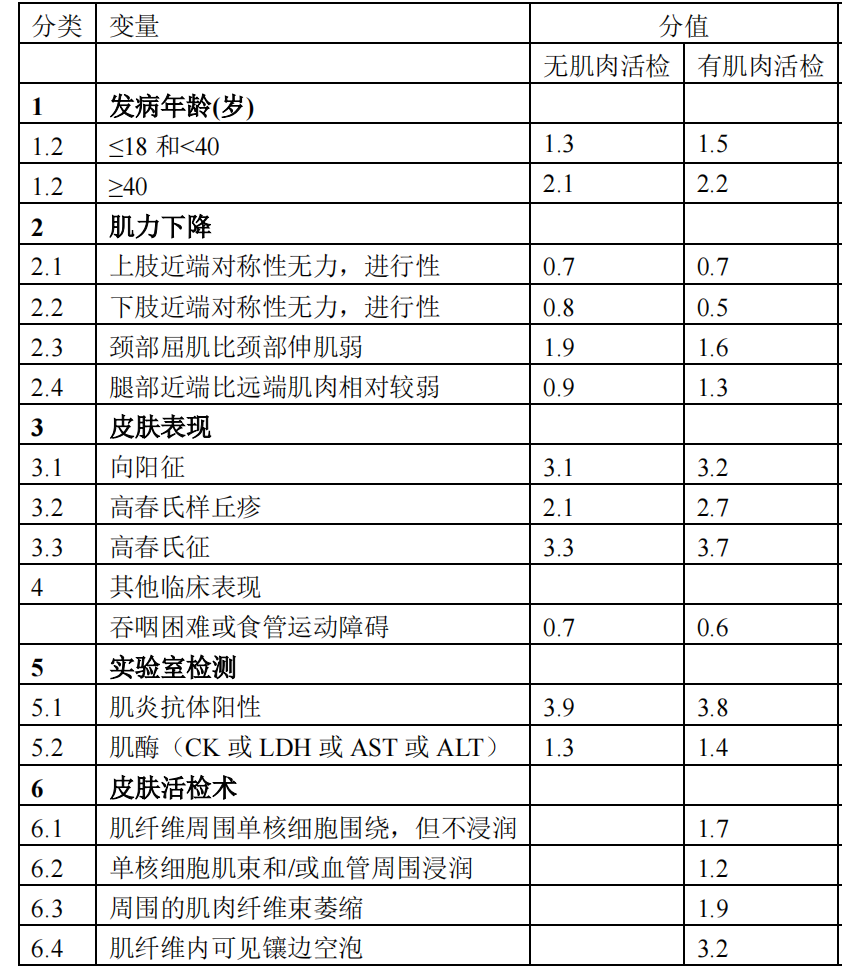
(Figure 1)

Discovery of Antibodies
Since the mid-1970s, one of the advances in idiopathic myositis research has been the discovery of myositis antibodies, including myositis-specific antibodies (MSA) and myositis-associated antibodies (MAA) (Figure 2). Myositis antibodies are of significant value in disease diagnosis, classification, and prognosis prediction. JDM antibodies are specific biomarkers, including anti-NXP2, TIF-1γ, MDA5, Mi2, and SAE antibodies (Figure 3). These antibodies have been found to be closely related to the clinical features, disease progression, treatment response, and long-term prognosis of JDM; assessing JDM disease status and treatment response is particularly important.
Anti-NXP2 antibodies are antibodies targeting nuclear matrix protein 2 (NXP2). In JDM, anti-NXP2 antibodies are one of the most common (20-23%) antibodies in children.
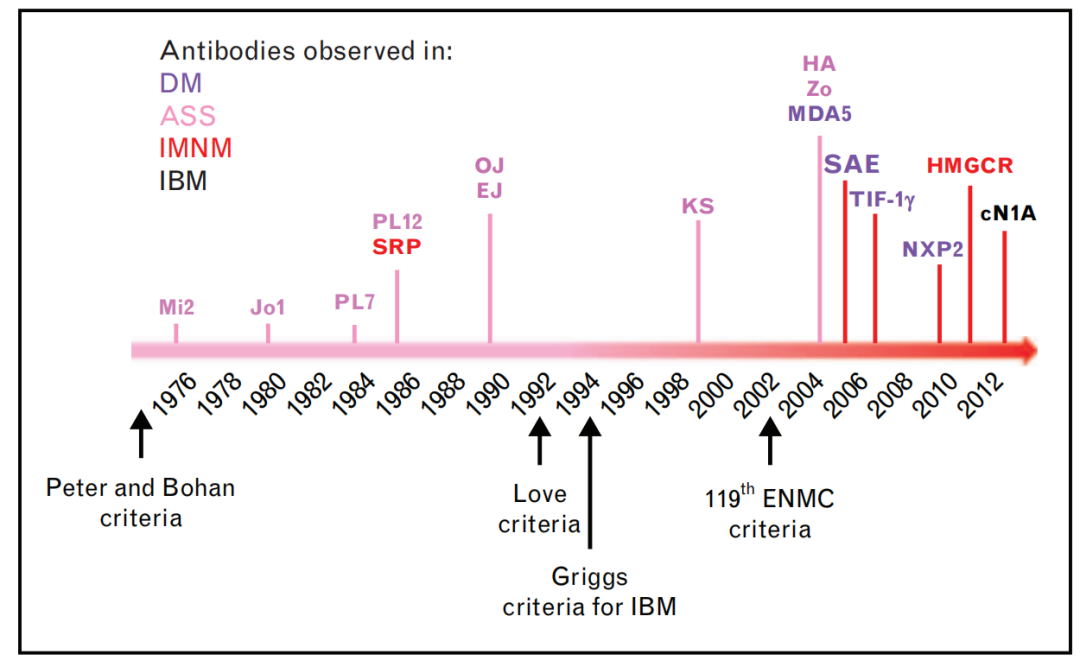
(Figure 2)
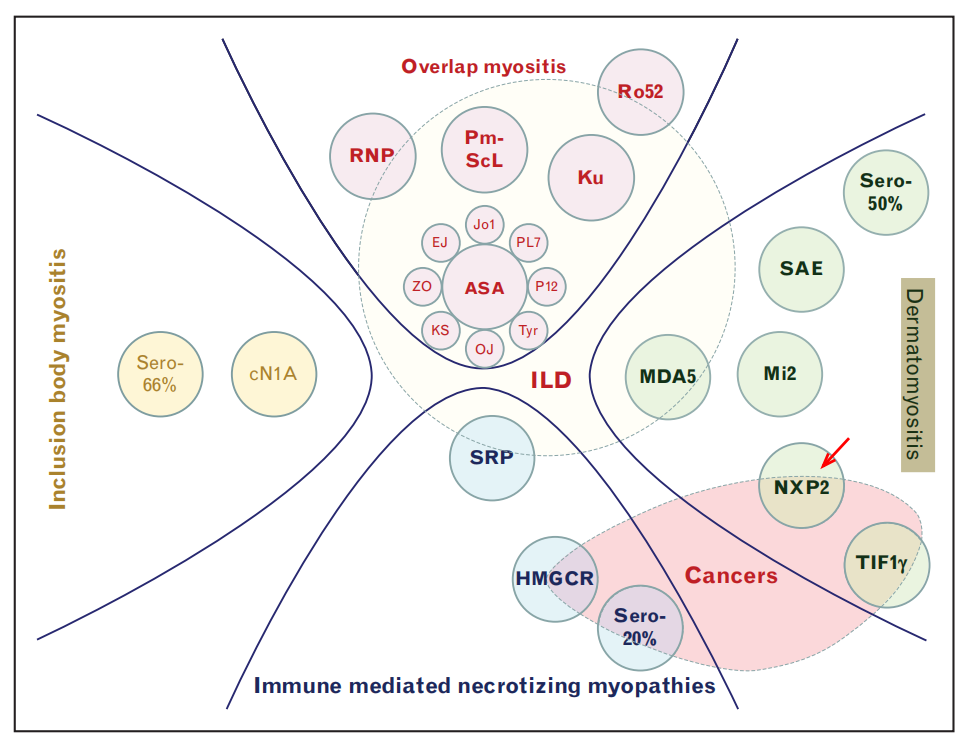
(Figure 3)

Clinical Features
NXP2 dermatomyositis is clinically characterized by typical rashes like heliotrope rash (see below), relatively severe muscle damage, showing significant muscle strength decline or muscle weakness, which can involve the pharyngeal muscle group and gastrointestinal smooth muscle, leading to dysphagia, gastrointestinal vasculitis (vomiting, abdominal pain, etc.). Severe cases can cause aspiration and gastrointestinal perforation, which are one of the causes of patient mortality. Patients who do not receive timely treatment or respond poorly to treatment may experience muscle atrophy and paralysis, skin ulcers, and soft tissue calcification (Figure 4).
Early accurate diagnosis and rapid establishment of a reasonable treatment plan in the rheumatology department are crucial, as it not only helps improve the survival rate of children but also allows them to achieve a good quality of life.
However, due to the rarity of JDM as a childhood disease, the public’s lack of understanding of this condition, limited expression ability in children, and diverse clinical symptoms, some children’s early symptoms may be overlooked, or they may be repeatedly referred among ophthalmology, dermatology, neurology, hepatology, etc., without receiving timely diagnosis and correct treatment, resulting in delayed treatment.
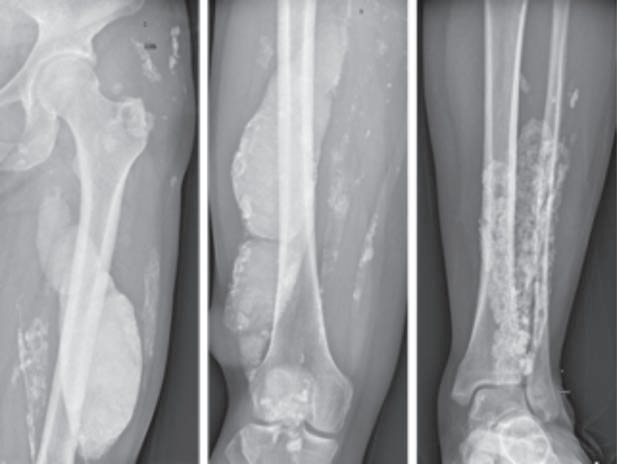
(Figure 4)

Early Symptoms Are Key
So how can NXP2 dermatomyositis be detected, diagnosed, and treated early?
1.Pay Attention to Skin Manifestations: One early symptom of dermatomyositis in children is skin rashes. Typical skin changes, including purplish-red skin on the eyelids accompanied by periorbital edema (heliotrope rash, Figure 5); red scaly rashes on the extensor surfaces of the metacarpophalangeal joints, proximal interphalangeal joints, elbows, and knees (Gottron’s papules, Gottron’s sign, Figure 6); as well as facial rashes, neck rashes (V-sign rashes), and back neck rashes (shawl sign). Children with NXP2 positive JDM particularly exhibit prominent periorbital edema or “swelling.” It is hoped that this skin window can be used for early diagnosis and timely screening for inherent lesions of JDM.
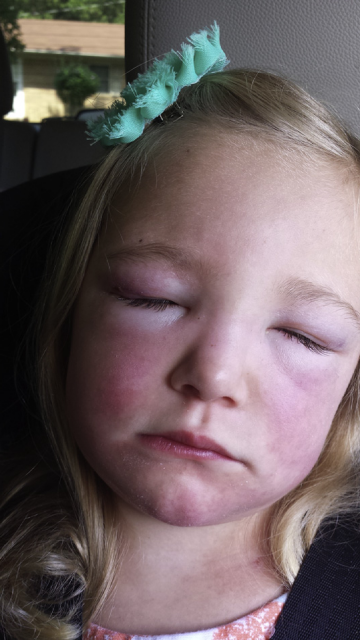
(Figure 5, image source from literature)
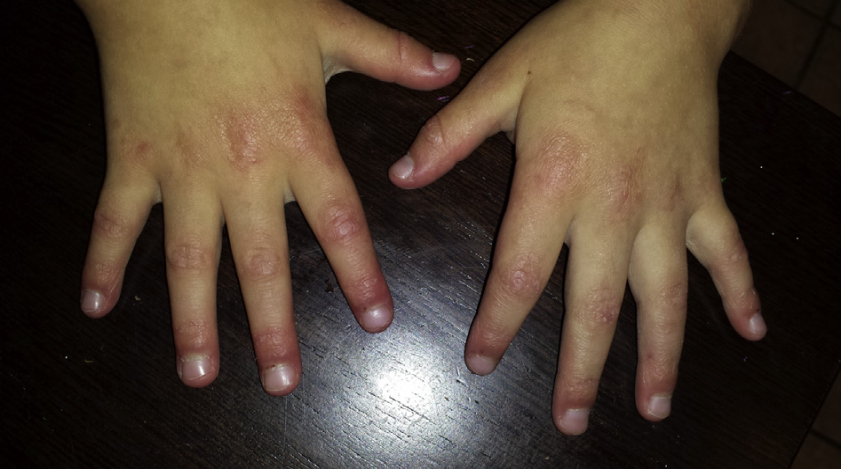
(Figure 6)
2. Pay Attention to Child’s Activity Level: The characteristic of muscle damage in dermatomyositis is symmetrical and proximal muscle weakness, which worsens progressively. Involvement of neck muscles can lead to an inability to lift the head; upper limb muscles may manifest as an inability to raise the arms, comb hair, or wash the face; lower limb muscles may show an inability to walk long distances, needing to sit down to rest after walking just a hundred meters, difficulty climbing stairs, or inability to stand up after sitting or squatting; if the pharyngeal muscles are involved, it can present as hoarseness and difficulty swallowing; becoming a “lazy child.” In addition to muscle weakness, children may also experience muscle pain or tenderness. Parents should be aware that when a child exhibits these symptoms, they should consult a rheumatologist for a professional assessment rather than attributing it directly to “laziness.”

(Illustrative image, image source from the internet)
3. Do Not Ignore Auxiliary Examinations: JDM is often accompanied by elevated muscle enzymes and liver enzymes, and may also show interstitial lung disease. Therefore, when laboratory indicators show elevated muscle enzymes or liver enzymes for unknown reasons, or lung imaging suggests interstitial lung disease, it is recommended to promptly consult a rheumatology specialist to rule out this condition and avoid delaying treatment.
4. Pay Attention to Myositis Antibody Testing: For children diagnosed with JDM for the first time, we recommend comprehensive myositis antibody testing, which can help clarify the diagnosis and differential diagnosis of the disease while providing important information regarding disease subtypes, predicting clinical phenotypes, and prognosis; it also serves as a reference for doctors in formulating treatment plans (drug selection, treatment intensity). Below is Table 1 (the relationship between myositis-associated autoantibodies and clinical phenotypes and prognosis of juvenile idiopathic inflammatory myopathy). Additionally, for NXP2 positive juvenile dermatomyositis, monitoring changes in NXP2 titers can reflect disease activity and help doctors make corresponding adjustments in treatment.
Table 1 (The Relationship Between Myositis-Associated Autoantibodies and Clinical Phenotypes and Prognosis of Juvenile Idiopathic Inflammatory Myopathy)
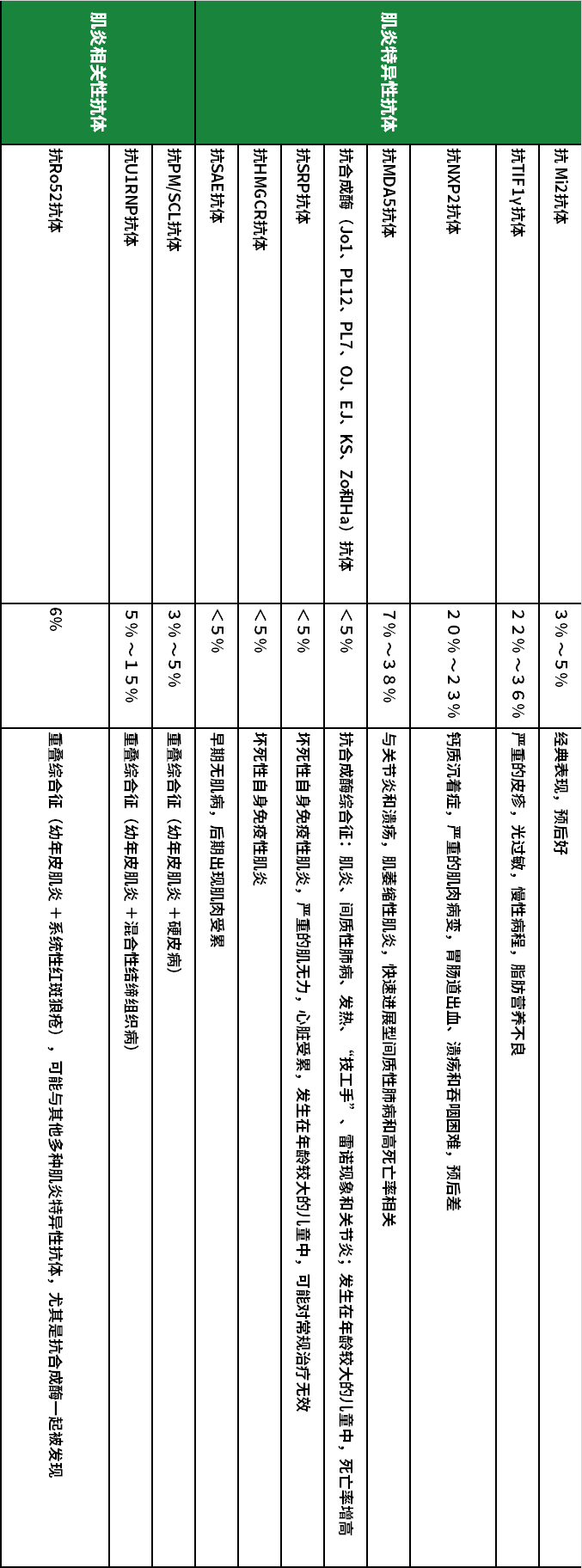
(Table 1, click to view the large image horizontally)

This article is almost at an end. We hope that through this article, parents and other specialists in the field can gain a simple understanding of NXP2 positive JDM, avoiding misdiagnosis and missed diagnosis. We hope that all parents can find the right department for early diagnosis and treatment from specialists to avoid misdiagnosis.
References:
1) Chinese Medical Association Pediatrics Branch Rheumatology Group, et al. Expert Consensus on the Diagnosis and Treatment of Juvenile Dermatomyositis [J]. Chinese Journal of Practical Pediatrics, 2022, 37(10):726-732.
2) Yang H, Lu X, Peng Q, et al. Differential Clinical Associations of Anti-Nuclear Matrix Protein 2 Autoantibodies in Patients With Idiopathic Inflammatory Myopathies. Arthritis Rheumatol. 2018;70 (8):1288-1297. doi:10.1002/art.40491
3) Benveniste O, Stenzel W, Allenbach Y. Advances in serological diagnostics of inflammatory myopathies. Curr Opin Neurol. 2016;29 (5):662-73.doi:10.1097/WCO.0000000000000376
4) Lundberg IE, Tjärnlund A, Bottai M, et al. 2017 European League Against Rheumatism/American College of Rheumatology classification criteria for adult and juvenile idiopathic inflammatory myopathies and their major subgroups. Ann Rheum Dis. 2017;76 (12):1955-1964. doi:10.1136/annrheumdis-2017-211468
Author of the Article

Reviewed by: Sun Li, Zeng Qiaoqian, Guan Wanzhen
Supported by: SLE Relief Grocery Store
