Key Components and Mechanisms of ADC
-
Stability
The instability of linkers in the bloodstream (also known as premature payload release) is a major factor limiting the therapeutic index of ADCs. Stabilization technologies are strategies to improve the therapeutic window. If the drug cannot be linked to the ADC, it cannot be delivered directly to tumor cells via antibodies, and free drugs can exacerbate systemic toxicity. However, this view oversimplifies the issue, as it overlooks the distribution of ADCs in normal tissues; in reality, only a small portion of the administered dose reaches the tumor site.
-
Types of Linkers
The mechanisms of traditional ADCs include target binding, internalization, and degradation, ultimately releasing cytotoxic drugs. Linkers that can directly release unmodified drugs are termed “cleavable,” while those that require complete degradation of the ADC to release modified payloads are termed “non-cleavable.” Most payloads become inactive after structural modification, making the former more of a focus. Cleavable linkers are typically classified by their cleavage pathways (proteolytic, reductive, pH, glycosidic hydrolases, etc.). Tumor-selective cleavable linkers have only been evidenced in certain in vitro studies. Currently, all approved ADCs utilize cleavable linkers, with the exception of trastuzumab emtansine.
-
Types of Instability
Linker instability can be divided into two categories:
-
Linker-drug instability: The drug is released prematurely before reaching the target.
-
Antibody-linker instability: The entire linker-drug complex dissociates from the antibody.
Most cleavable linkers exhibit some degree of linker-drug instability, as they can release drugs through chemical or enzymatic cleavage, although the extent of cleavage in the bloodstream varies. Antibody-linker instability is related to the conjugation structure. For example, lysine conjugation via amide bonds is very stable, while cysteine conjugation via maleimide shows significant variability in stability.
-
Stability of Approved ADCs
Aside from the inherent clearance mechanisms of ADCs, all currently approved ADCs exhibit some form of linker instability in the bloodstream (either antibody-linker or linker-drug instability, or both). The following diagram (A) illustrates the stability of approved ADCs in the circulatory system, with percentages indicating the estimated amount of payload drug still conjugated to the antibody after 7 days of incubation in plasma.
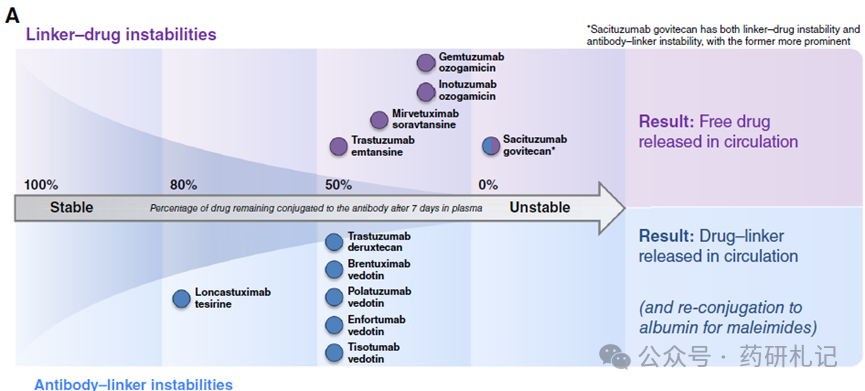
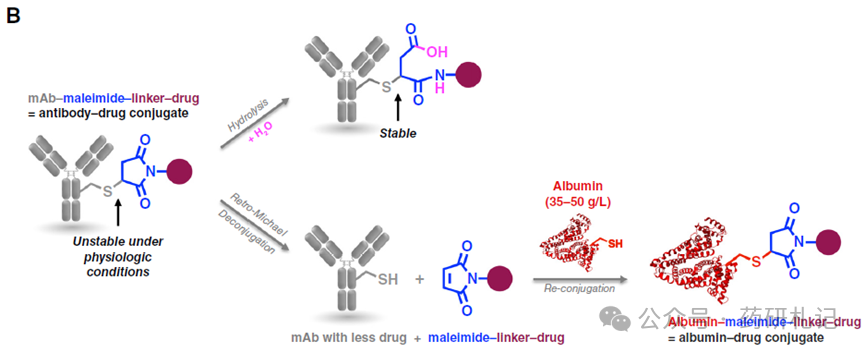
Seven approved ADCs utilize thiol-maleimide (cysteine) conjugation, while four use lysine conjugation. For thiol-maleimide ADCs, the hydrolysis of maleimide and the reverse Michael addition reaction are competing processes, with the former stabilizing the antibody-linker bond and the latter releasing the maleimide-containing linker (Figure B). The balance of these two processes is influenced by the chemical structure of the linker and the microenvironment of the cysteine residues.
In the bloodstream, the maleimide hexanoic acid (MC) linker used by brentuximab vedotin, polatuzumab vedotin, enfortumab vedotin, tisotumab vedotin, and T-DXd dissociates by about 50% within a week. The 4-maleimido methyl cyclohexane-1-carboxylic acid (MCC) linker used by sacituzumab govitecan (SG) has a similar dissociation rate to MC. The shorter maleimide propionic acid (MP) linker used by loncastuximab tesirine exhibits lower antibody-linker instability, with about 20% of the drug-linker lost within a week.
Most cysteine-conjugated ADCs have higher antibody-linker instability than linker-drug instability. An exception is SG, whose highly unstable carbonate rapidly decomposes in the bloodstream (half-life of about 24 hours), releasing free SN38 drug. Maleimide drug-linkers dissociated from antibodies can transfer to other thiol-containing biomolecules (primarily human serum albumin, with serum concentrations of 35-50 mg/mL). Although albumin has a half-life of about 3 weeks in the human body, and maleimide dissociation forms albumin-drug conjugates, the metabolism of such conjugates in patients remains unclear. Preclinical studies have sufficiently demonstrated the tumor delivery potential of albumin-cytotoxic conjugates, suggesting that similar uptake may occur in patients.
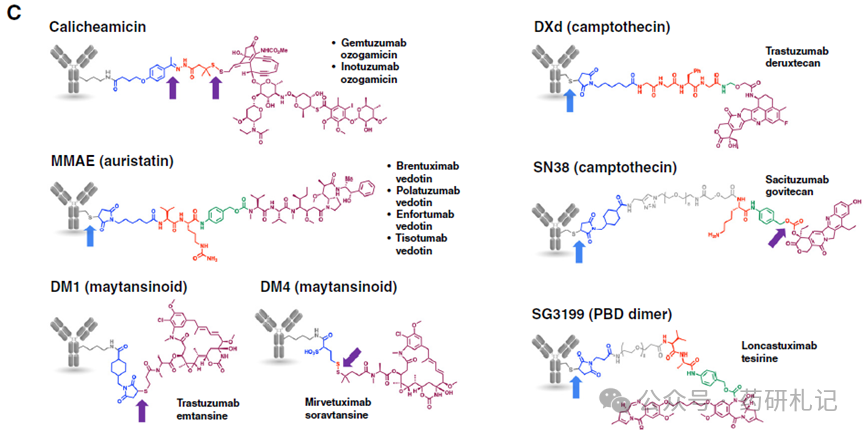
For ADCs with linker-drug instability, different chemical groups are responsible for the release of free drugs (Figure C), including disulfide bonds, hydrazone bonds, carbonates, and thiol-maleimides.
ADC Stabilization
Theoretically, increasing the stability of the antibody-linker and linker-drug in the bloodstream could reduce free drugs, lower toxicity, and enhance efficacy. However, this understanding of the ADC therapeutic window overlooks two key points:
-
Free drugs can reach pharmacologically relevant blood concentrations and may significantly contribute to anti-tumor effects.
-
Stabilization may increase normal tissue exposure to the conjugated drug ADC.
The tissue distribution and metabolic sites of ADCs have a decisive impact on their toxicity profile, especially when each antibody carries more payloads (such as stabilized linker ADCs), as the vast majority of the injected dose is metabolized in normal tissues rather than tumor sites. Although the disposal of ADCs depends not only on stability, stabilization may lead to more payloads being delivered to specific organs based on preferred metabolic sites (i.e., targeting).
Various antibody-linker stabilization methods have been clinically evaluated, with the most notable being site-specific conjugation technology. Compared to traditional ADCs, site-specific conjugated ADCs may increase the amount of bound drug exposure (indicating improved stability and biophysical properties), but the maximum tolerated dose (MTD) of the drug remains nearly unchanged. These stabilization methods often reduce blood toxicity, but other tissue-specific dose-limiting toxicities may emerge, possibly due to increased accumulation of high drug-load ADCs in normal tissues.
For example, the site-specific conjugated DAR2 CD79b vedotin ADC DCDS0780A exhibited a higher amount of bound drug exposure (AUC0-21 approximately 4 times) compared to DAR4 polatuzumab vedotin, yet both had similar free MMAE exposure levels. The standardized cytotoxic content of DCDS0780A was comparable to polatuzumab vedotin and other vedotin ADCs (site-specific or random conjugation). Despite showing advantages in preclinical studies, DCDS0780A encountered significant off-target toxicity—ocular toxicity—clinically, while polatuzumab vedotin has not reported such effects. Ultimately, the less stable polatuzumab vedotin was approved, while DCDS0780A was discontinued in development.
Considering the complexity of ADC pharmacology and the intertwining of multiple variables, it is challenging to isolate the impact of stability assessment. Ideally, clinical data comparing ADCs with identical targets, antibodies, linkers, and payloads within the same patient population would be analyzed.
To date, over 30 site-specific conjugated ADCs have been terminated in clinical development, with no site-specific conjugated ADCs approved. It is evident that site-specific technology and/or fully stable linkers are not the ultimate solution to the challenges faced by ADCs, but they remain valuable tools for generating drugs with diverse characteristics that may be approved in the future.
References:
Raffaele Colombo, Paolo Tarantino, Jamie R. Rich, Patricia M. LoRusso, Elisabeth G.E. de Vries; The Journey of Antibody–Drug Conjugates: Lessons Learned from 40 Years of Development. Cancer Discov 1 November 2024; 14 (11): 2089–2108