
Challenges in Diagnosis and Treatment: An International Stakeholder Perspective
Compiled/Translated by Chen Kang
Background
Severe Primary Insulin-like Growth Factor-I (IGF-I) Deficiency (SPIGFD) is a rare growth disorder characterized by short stature (standard deviation score [SDS] ≤ 3.0), low circulating IGF-I (SDS ≤ 3.0) levels, and normal or elevated growth hormone (GH) levels. Laron syndrome is the most characteristic form of SPIGFD, caused by a defect in the GH receptor (GHR) gene. However, awareness of SPIGFD remains low, and patients continue to face challenges related to diagnosis, treatment, and care.
Objective
The aim of this report is to gather perspectives from multiple stakeholders on the main challenges faced by individuals and families affected by SPIGFD. By highlighting the critical gaps in awareness, diagnosis, and management of SPIGFD, this report intends to provide recommendations to improve care for SPIGFD patients globally.
Methods
An international group composed of clinical experts, researchers, and representatives from the SPIGFD community attended a half-day virtual meeting to discuss key unmet needs and opportunities to improve care for SPIGFD patients.
Results
As a rare disease, healthcare professionals (HCPs) have limited awareness and understanding of SPIGFD, posing significant challenges for patient diagnosis and treatment. Patients often face difficulties related to obtaining a formal diagnosis, delays in the initiation of treatment, and limited access to appropriate therapies. This has a considerable impact on patient health and quality of life, highlighting the need for more education and clearer guidance for healthcare providers. Support from patient advocacy groups is valuable in helping patients and their families find appropriate care. However, there is still a need for better understanding of the burden SPIGFD imposes on individuals beyond height, including impacts on physical, emotional, and social well-being.
Conclusion
To address the challenges faced by individuals and families affected by SPIGFD, the healthcare community needs to raise awareness of SPIGFD and reach a consensus on best practices for the care of affected individuals. Continued efforts are needed globally to challenge existing perceptions of SPIGFD and identify solutions that promote equitable access to appropriate care.
Introduction
Severe Primary Insulin-like Growth Factor-I (IGF-I) Deficiency (SPIGFD) is a rare growth disorder that falls under the broader category of IGF-I deficiency. SPIGFD is characterized by severe short stature, low circulating IGF-I levels, and normal or elevated growth hormone (GH) levels. A simple overview of the SPIGFD disease pathway is illustrated in Figure 1; more details regarding the molecular pathways involved in IGF-I signaling have been described elsewhere (e.g., Puch and Castilla-Cortázar [2012]; Argente et al. [2017]). The global prevalence of SPIGFD remains uncertain, partly due to varying definitions of the condition. In the European Union (EU), it is estimated that 2 in every 10,000 individuals are affected by primary IGF-I deficiencies (PIGFD); as a subclass of PIGFD, it is speculated that fewer individuals are affected by SPIGFD. A cohort study in France indicated that approximately 0.8%-1.2% of children referred to pediatric endocrinology due to suspected linear growth retardation were diagnosed with SPIGFD. Recombinant human IGF-1 (rhIGF-1) is currently the only effective treatment for SPIGFD patients.
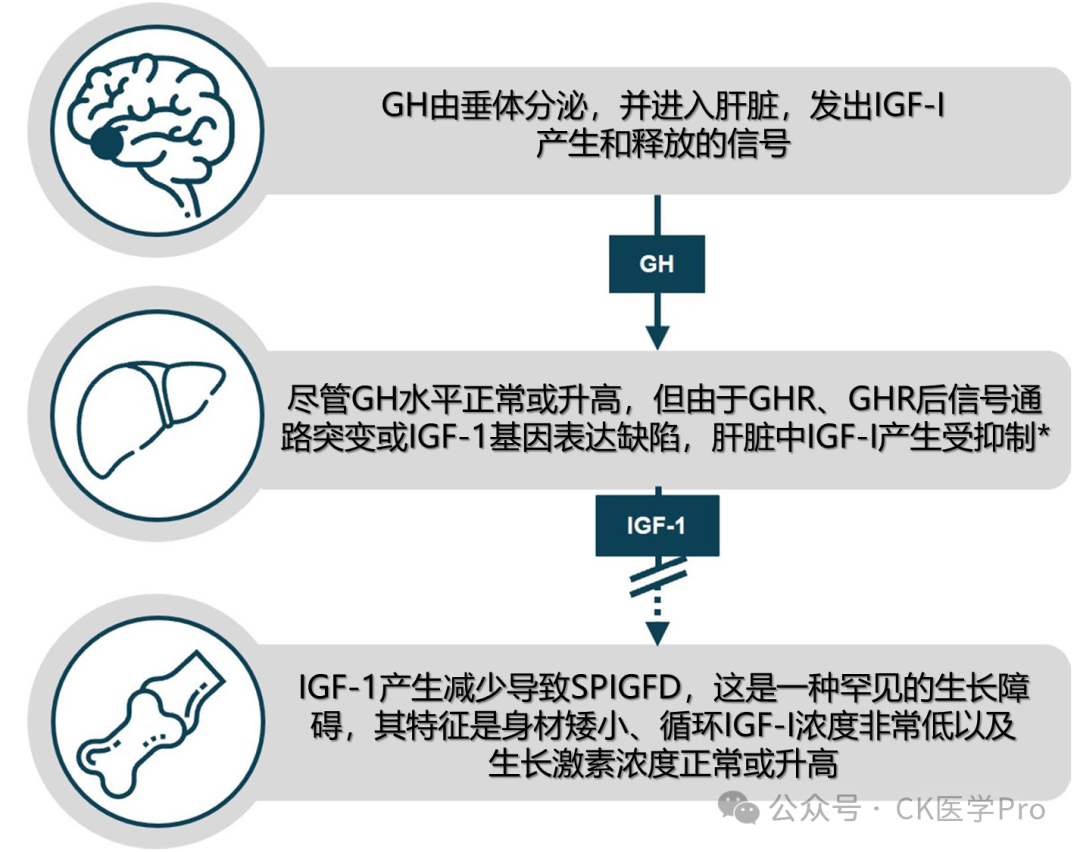
SPIGFD can be caused by key gene mutations in the GH-IGF axis, such as mutations in the genes encoding the GH receptor (GHR), signal transducer and activator of transcription 5B (STAT5B), IGF-I, or acid-labile subunit (ALS) (Table 1). The most typical form of SPIGFD is Laron syndrome, caused by mutations in the GHR gene. European registry data emphasize that IGF-I deficiency may have multiple genetic causes, but it remains unclear that in the EU Increlex® growth forum database, confirmed GHR deletions or mutations account for only 13% of children receiving rhIGF-1. Additionally, milder forms of the disease may still go undetected. Therefore, the diagnostic pathway for SPIGFD is currently challenging, and the exact genotype distribution remains uncertain.
|
Affected Gene |
Details |
|
IGFALS |
Deficiency in the IGFALS gene leads to decreased serum ALS associated with moderate short stature, delayed puberty, and reduced serum IGF-I. |
|
GHR |
The genetic defect in the GHR gene is the underlying cause of the typical form of SPIGFD known as Laron syndrome. |
|
IGF-I |
Homozygous mutations in IGF-I are very rare and are associated with severe short stature, deafness, and insulin resistance. |
|
IκBα |
Mutations in IκBα are associated with short stature and immune deficiency. |
|
PAPPA2 |
Defects in PAPPA2 disrupt the release of circulating IGF-I, leading to varying degrees of short stature and insulin resistance. |
|
PTPN11 |
Activation of PTPN11 mutations leads to dephosphorylation of STAT5B, resulting in downregulation of its activity and partial GH insensitivity. |
|
STAT5B |
Mutations in STAT5B can lead to short stature and severe immune dysfunction. |
Source: Wit et al. 2012; Hwa et al. 2021.
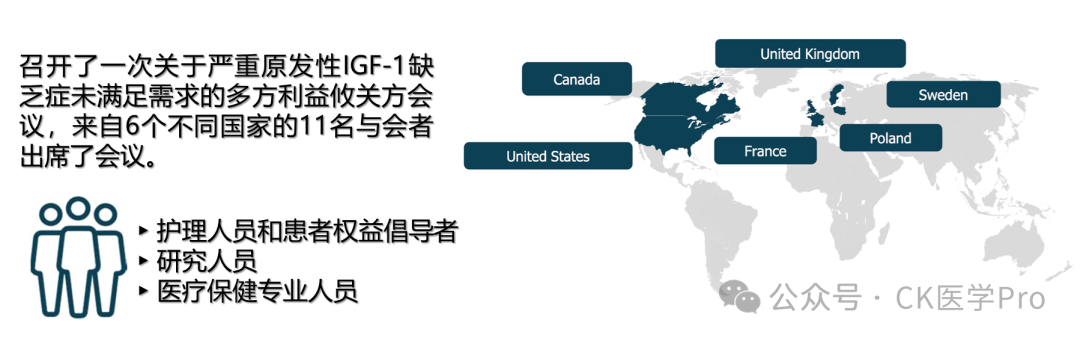
Evidence from a large cohort in Israel indicates that SPIGFD imposes a heavy burden on individuals and their caregivers, including challenges in navigating the diagnostic pathway and practical difficulties in achieving appropriate education, mobility, access to age-appropriate clothing, and providing ergonomic environments as they transition into adulthood. However, the non-growth-related impacts of SPIGFD are largely uncertain, and little is known about the overall impact of the disease on patients and their families globally. To gather perspectives on the broader challenges faced by SPIGFD patients, a multi-stakeholder meeting was convened, bringing together an international group of clinical experts, researchers, caregivers, and representatives from patient advocacy organizations. Utilizing their expertise and/or lived experiences, the group identified key challenges and highlighted opportunities and recommendations to help address unmet needs and improve care for SPIGFD patients. The main outcomes of the discussions are presented here.
Methods
A targeted literature review was conducted to identify evidence regarding the diagnosis, treatment opportunities, unmet needs, and best practices for the care of SPIGFD. The literature on the human, social, and economic burden of SPIGFD was also reviewed. The evidence collected was used to inform the discussion topics for the multi-stakeholder meeting.
The half-day virtual meeting included 11 participants, comprising clinicians, clinical nurse specialists, researchers, parents of children with SPIGFD, and representatives from patient advocacy organizations. The group included some internationally recognized experts and provided perspectives from six countries (Canada, France, Poland, Sweden, the United Kingdom, and the United States). Two experts in the field (Philippe Backeljauw and Mehul Dattani) co-chaired the meeting.
In accordance with best practices for patient involvement in research, standardized guidance for reporting participation of patients and the public (GRIPP) was utilized to report on patient and public involvement in the project.
Current Awareness
The main challenges and opportunities identified during the multi-stakeholder meeting are summarized in Figure 2.
It is estimated that the point prevalence of rare diseases globally is 3.5%-5.9%, cumulatively affecting 263-446 million people at any given time. However, awareness and understanding of specific rare diseases among healthcare providers and the broader community remain inadequate. For example, recent studies indicate that training and education regarding rare diseases among healthcare professionals are limited, with one study reporting that as many as 94.6% of clinicians believe their relevant knowledge is poor. As a rare disease, there is a widespread lack of awareness of SPIGFD within the healthcare community and beyond. Few healthcare professionals have firsthand experience caring for SPIGFD patients, and newly trained pediatric endocrinologists may lack the necessary exposure to recognize this condition. Moreover, much of the research and information dissemination regarding IGF-I deficiency has focused on the genetic details of diagnosis and treatment rather than the practicality of these processes. This has significant implications for the diagnosis and treatment of SPIGFD, as frontline HCPs may be unable to correctly diagnose the condition and refer patients to specialists. Introducing specialized medical education workshops focusing on the practicality of diagnosis and treatment would enhance awareness of SPIGFD and provide a platform to address unmet needs in SPIGFD care.

The general lack of awareness and understanding of SPIGFD also permeates more broadly, with limited information targeted at individuals outside the medical community. Access to comprehensive resources to understand SPIGFD is particularly important for patients and their families, providing tools to understand the complexities of SPIGFD and emphasizing the importance of early diagnosis and treatment adherence. Patient and caregiver advocacy groups, such as the MAGIC Foundation (USA) and the Children’s Growth Foundation (UK), play a key role in this regard, providing platforms for families to share their experiences and concerns via social media and conference networks. These groups also provide valuable support through educational materials for patients with growth disorders. However, there remains a continuous need for readily available information to enhance awareness and understanding of SPIGFD among affected individuals and the broader community.
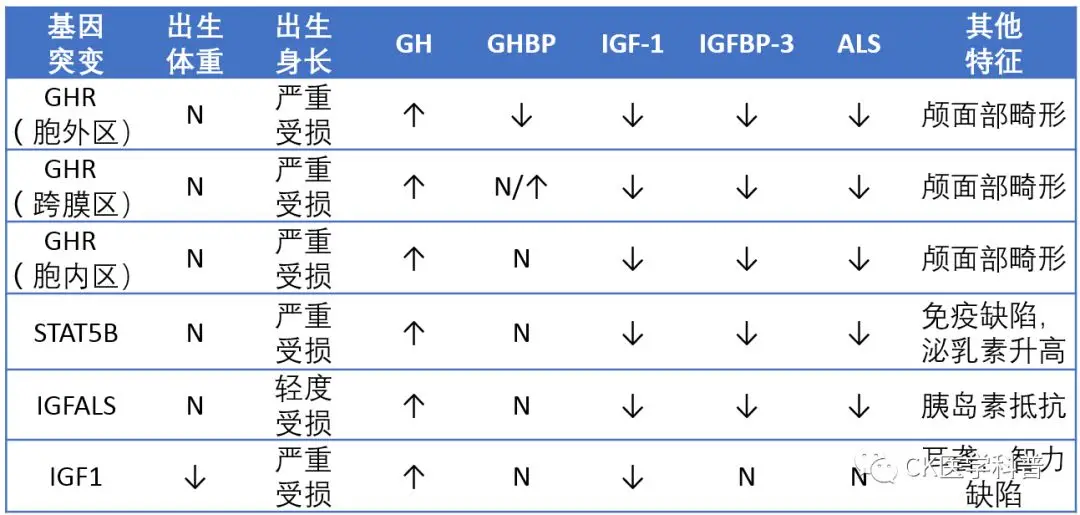
Diagnosis (Table S1)
Table S1 Gene Mutation Sites Leading to Laron Syndrome on the GH/IGF-1 Axis
And Their Clinical and Biochemical Features
Clinical Diagnosis
To diagnose SPIGFD, HCPs refer to a wide range of diagnostic criteria and examinations related to growth measurements. However, there are discrepancies in the diagnostic pathways published in different regions, and clinical standards for SPIGFD diagnosis vary. For example, due to differences in national growth charts, the diagnostic threshold of 3 SDS translates into different detection height values based on the specific country/region. This leads to potential variations in diagnostic standards and SPIGFD thresholds globally. For instance, growth charts in France, Sweden, and Italy indicate heights for 5-year-old boys of 97.0cm, 98.0cm, and 95.5cm, respectively. Therefore, it is crucial to use geographically appropriate and up-to-date growth charts. Additionally, some growth charts only provide percentiles, which require pediatric endocrinologists to manually calculate the SDS for children suspected of SPIGFD, making comparisons more challenging.
As part of a series of diseases with varying degrees of severity, the narrow margins of SPIGFD diagnosis may limit HCPs from considering less “classic” types of SPIGFD, including some autosomal dominant forms. This may delay diagnosis, thereby delaying the initiation of treatment and affecting treatment outcomes. In cases of familial short stature, comprehensive diagnostic evaluations must be conducted if short stature is inherited from one or both parents, not only to identify chronic, unrecognized diseases but also to confirm any possible cases of SPIGFD. Similarly, it is also important to conduct further IGF-I testing in children who respond poorly to stepwise GH treatment.
Supplement: Clinical Manifestations
1. Growth Disorders and Other Malformations
Children with Laron syndrome typically have normal birth weights (2500-4000g), but some may have birth lengths below 48cm (42-47cm). After birth, growth velocity slows significantly, with an average height of -4 to -10 SDS; untreated male patients have adult heights ranging from 116 to 142cm, while female patients range from 108 to 136cm. Additionally, patients with Laron syndrome have small hands, feet, and internal organs. Some patients may have other congenital malformations, such as hip dislocation, aortic valve stenosis, cryptorchidism, congenital cataracts, etc.
2. Facial Features
Characteristic facial changes in Laron syndrome patients include a prominent forehead, saddle nose, and small mandible, with a head-to-body ratio larger than that of normal children, but the absolute head circumference remains small compared to peers. Patients may have sparse hair, and some may exhibit the ‘sunset sign’; patients from the Mediterranean and Middle Eastern regions may show blue sclera. Delayed dental eruption, late loss of primary teeth, and multiple cavities are common, and dental malocclusion may occur due to a shorter mandible.
3. Musculoskeletal System
Some patients exhibit congenital hip dislocation and avascular necrosis of the femoral head. Due to midfacial skeletal underdevelopment and abnormal pharyngeal anatomy, patients may have a high-pitched voice. 85% of patients experience limited elbow joint mobility.
4. Metabolic Disorders
Patients with Laron syndrome often have metabolic abnormalities, including hypoglycemia in the neonatal and infant periods, with gradually worsening obesity in adulthood. However, studies on glucose metabolism in Laron syndrome patients remain controversial. Patients with Laron syndrome who are obese also exhibit hyperlipidemia.
5. Delayed Sexual Development
Patients with Laron syndrome have smaller genitals, with males exhibiting micropenis and small testes; ultrasound examinations in females reveal small ovarian volume. Most patients show delayed sexual development, with male patients experiencing testicular enlargement between 13-16 years, axillary hair growth around 16 years, and ejaculation between 17-21 years. Female patients typically have their first menstruation between 13-15 years. Although sexual development is delayed, both male and female patients can eventually develop mature sexual function and fertility.
6. Tumor Incidence
The risk of tumor occurrence in patients with Laron syndrome is significantly lower than that of the general population.
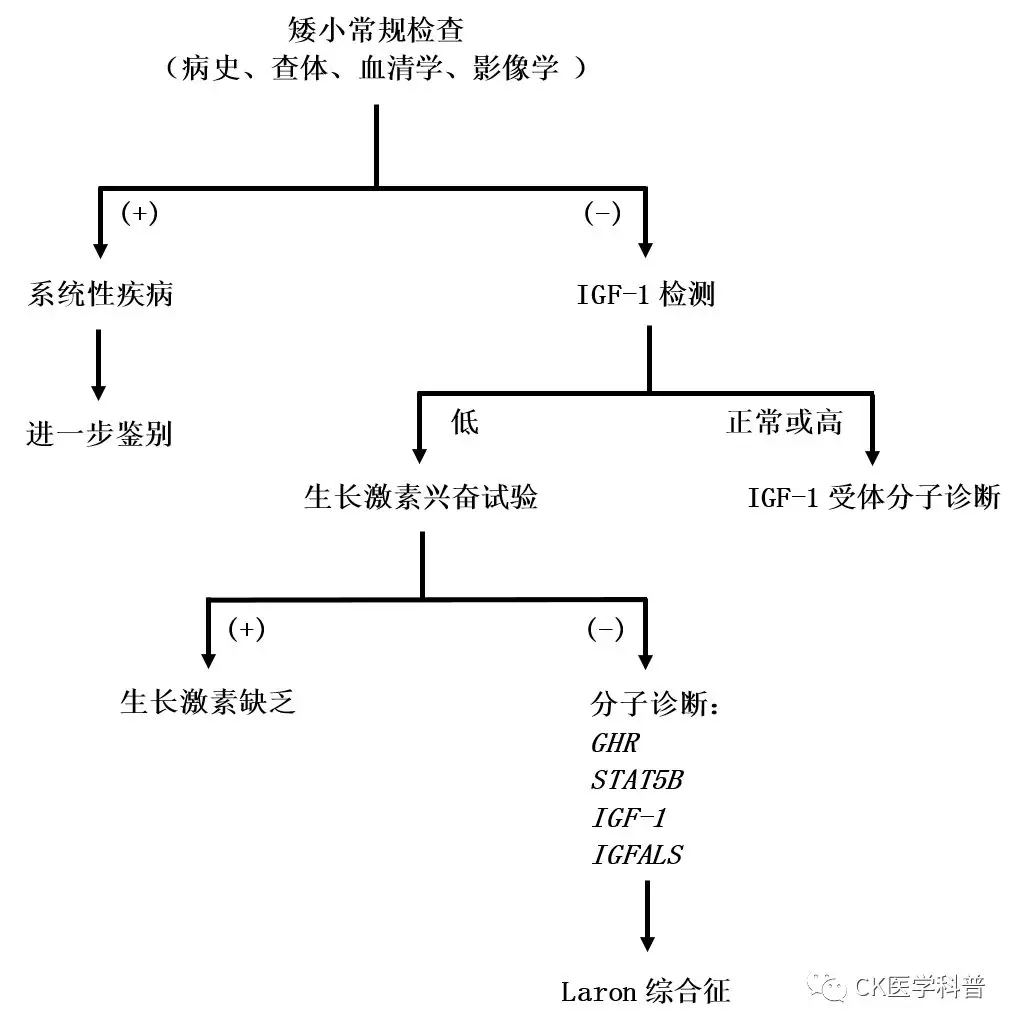
Biochemical Diagnosis
Biochemical standards are typically necessary to support the comprehensive diagnosis of SPIGFD in this heterogeneous patient population. Detection of IGF-I deficiency usually includes measuring serum IGF-I and demonstrating normal or increased GH secretion. However, there are concerns about the sensitivity of commercially available tests for detecting IGF-I concentration, which depends on a range of factors including age, sex, pubertal stage, nutritional status, and genetic factors.
Additionally, there is a lack of normative data regarding IGF-I SDS and percentiles, and limited data exist for the youngest pediatric patients, leading to a wide range of ‘normal’ values, complicating biochemical diagnosis of SPIGFD. While published biochemical testing recommendations exist, clinicians appear to have limited adoption of these recommendations, resulting in a lack of international standards for sample collection/storage procedures. This leads to discrepancies in hormone concentration estimates and clinical interpretations across different testing methods. Furthermore, diagnostic criteria related to serum IGF-I concentrations also vary by region. For example, in the U.S., the threshold for diagnosing SPIGFD is an IGF-I concentration of SDS ≤ 3.0, while in the EU it is <2.5 SDS points.
Although not a necessary requirement for the diagnosis of SPIGFD, IGF-I generation testing can also be utilized in clinical practice to investigate GH secretion and sensitivity, thus supporting the diagnostic process. However, the lack of normative reference data means that the results of this test are rarely definitive, unless extreme GH insensitivity is typically present in classic SPIGFD cases. Therefore, conducting such tests in non-classic SPIGFD cases may lead to prolonged and unnecessary evaluations, imposing a considerable economic and resource burden on healthcare services.
Supplement: Diagnostic Process
Accessing Treatment
Once diagnosed with SPIGFD, treatment with rhIGF-1 should be initiated as soon as possible. However, discussions emphasize that many individuals receive treatment at unacceptably late stages due to delays or misdiagnosis, compounded by limited awareness among healthcare providers regarding SPIGFD. Given that the height SDS of affected individuals continues to decline with age, patients with early heights below 3 SDS will ultimately experience more severe short stature than older patients. Therefore, early intervention is particularly important for children with SPIGFD, as it can optimize long-term treatment outcomes, thereby alleviating the burden of short stature in adulthood.
Access to rhIGF-1 varies by geographical region, depending on regulatory, political, and broader socioeconomic contexts. Although rhIGF-1 was developed in 1986, it was not approved for use by the U.S. FDA until 2005, and specific coding for its prescription was not provided until 2021. In Europe, the European Medicines Agency (EMA) approved the marketing authorization for this therapy under exceptional circumstances in 2007. In Canada, rhIGF-1 was not approved until 2021. In regions where rhIGF-1 is unlicensed, such as Australia, patients may seek treatment through managed access programs. However, for other regions, including much of South America, the Middle East, and Africa, SPIGFD patients are unable to access rhIGF-1 locally due to lack of approval. Weak infrastructure, political turmoil, economic challenges, or other barriers to adequate patient safety follow-up may further hinder access for these patients, leading to the unexpected withdrawal or suspension of managed access programs that do exist in these regions. These challenges are pertinent to Middle Eastern countries, where SPIGFD cohort characteristics are well-defined, yet many patients come from low socioeconomic backgrounds.
To encourage the global approval of SPIGFD treatments, the need to enhance understanding of SPIGFD epidemiology and treatment responses remains unmet, particularly in areas where the characteristics of SPIGFD are less well-defined. Increased utilization of patient registries for data collection could facilitate this effort. Recognition of rhIGF-1 by major international healthcare organizations, such as the World Health Organization (WHO), could also support broader authorization and approval. Simultaneously, introducing well-managed compassionate use programs may help address challenges of inequitable access for SPIGFD communities in regions where treatment is currently unavailable.
Medical Education
Identifying and diagnosing SPIGFD patients is an active process that requires specific training and expertise from HCPs. However, in practice, HCPs often lack the necessary understanding to identify the classic features of Laron syndrome, let alone the atypical phenotypes of IGF-I deficiency (e.g., heterozygous STAT5B mutations). As a result, many SPIGFD cases either remain undiagnosed or are generalized under broader idiopathic short stature.
To improve clinical diagnoses, it is recommended that healthcare providers (including primary care physicians and pediatric endocrinologists) receive specific education and exposure to accept classic diagnostic evaluations for short stature, particularly SPIGFD. This should include detailed patient histories and information about appropriate examinations. Furthermore, training should be provided to HCPs to recognize the classic SPIGFD phenotype (Table 2) and genotype (Table 1), as this may help enhance understanding of non-classic and less severe cases of IGF-I deficiency. Training courses or programs specifically targeting primary endocrinologists will provide the necessary background knowledge to appropriately diagnose IGF-I deficiency patients. Training materials for healthcare providers should be made available in multiple languages to ensure equitable access and dissemination of resources internationally.
|
Clinical Features |
Details |
|
Growth |
Normal birth weight and length |
|
Growth retardation at birth |
|
|
Height deviation associated with low serum IGF-I |
|
|
Delayed bone age |
|
|
Small hands and/or feet |
|
|
Facial Features |
Sparse hair before age 7; receding hairline at all ages |
|
Prominent forehead |
|
|
Head disproportionately large due to short stature |
|
|
25% of patients under 10 years exhibit the ‘sunset sign’ (sclera visible above the iris at rest) |
|
|
Low nasal bridge, shallow orbits |
|
|
Reduced vertical facial dimensions |
|
|
Blue sclera |
|
|
Delayed retention of primary teeth; permanent teeth normal but crowded |
|
|
Unilateral ptosis, facial asymmetry |
|
|
Musculoskeletal Composition |
Decreased muscle mass, delayed walking |
|
Avascular necrosis of the femoral head (25%) |
|
|
High-pitched voice |
|
|
Thin skin, premature aging |
|
|
Limited elbow extension after age 5 |
|
|
Low-normal BMI in childhood, developing into high BMI in adulthood |
|
|
Significantly reduced ratio of lean body mass to fat compared to normal individuals |
|
|
Cognitive Function |
Impact on brain growth and structure |
|
Decline in cognitive function and motor performance |
|
|
Metabolic Characteristics |
Hypoglycemia |
|
Increased cholesterol and low-density lipoprotein-C |
|
|
Reduced sweating |
|
|
Insulin resistance |
|
|
Sexual Development |
Small genitals in childhood males |
|
Delayed puberty |
|
|
Normal fertility |
|
|
Other Functions |
Deafness |
|
Immune deficiency or severe immune dysfunction |
Source: Guevara-Aguirre et al. 1991; Laron 2001; Webb et al. 2012; Higashi et al. 2019; Cohen et al. 2014; Backeljauw et al. 2010; Bang et al. 2012; Ascenzi et al. 2019.
The list of non-growth effects of SPIGFD is not exhaustive; currently, research on the non-growth effects of SPIGFD is limited, and little is known about these studies.
Linking SPIGFD with other short stature syndromes associated with IGF-I deficiency or partial GH insensitivity (such as Silver-Russell syndrome or Noonan syndrome) provides a good starting point for the medical community to establish awareness and understanding of this rare disease. For example, the typical clinical features of Noonan syndrome include short stature (which may or may not be associated with low IGF-I concentrations), craniofacial features, and cardiac defects, which may lead to misdiagnosis of SPIGFD and vice versa. However, unlike SPIGFD patients, the recommended first-line treatment for Noonan syndrome is recombinant human growth hormone (rhGH). Further testing is needed to better describe the spectrum of short stature syndromes, as well as additional training to support HCPs in recognizing the key clinical nuances between these diseases to ensure timely and accurate diagnosis.
There is also a need for training of HCPs regarding the expected benefits of rhIGF-1 treatment for SPIGFD. Given that first-year height velocity data indicate that rhIGF-1 has a less pronounced impact on SPIGFD patients compared to rhGH on patients with growth hormone deficiency (rhIGF-1: 6.9 [6.5, 7.2] cm/year [n = 144] vs rhGH: 8.67 [7.5, 9.9] cm/year [n = 465]), some HCPs may perceive the benefits of rhIGF-1 for SPIGFD as insufficiently significant. However, this should not constitute a reason to disregard treatment; an increase in height velocity of 6.9 cm/year can still provide meaningful benefits for SPIGFD patients. Training to communicate this to healthcare providers will be valuable. Such training should also reassure healthcare providers that the potential benefits of rhIGF-1 often outweigh the risks of side effects, particularly hypoglycemic events. Hypoglycemia is the most common adverse event (AE) associated with untreated IGF-I deficiency, and without proper management, rhIGF-1 treatment may worsen it; studies in Poland and the U.S. estimate that 7%-42% of SPIGFD patients treated with rhIGF-1 experience hypoglycemia. This may lead to hesitancy among HCPs when prescribing rhIGF-1. However, other studies indicate that rhIGF-1 treatment does not increase the incidence of hypoglycemic events, and such events can be effectively prevented through proper nutrition and monitoring. Additionally, the frequency of hypoglycemic AEs reported in clinical trials (49%) is significantly higher than post-marketing data (28%), suggesting that this risk can be controlled with adequate blood glucose monitoring. It is recommended to monitor capillary blood glucose when initiating rhIGF-1 treatment until a well-tolerated dose is achieved. Case studies involving children treated with rhIGF-1, including data on adult height and hypoglycemic events, could serve as important educational tools for HCPs to share experiences and best practices.
Perspectives on SPIGFD
The primary goal of rhIGF-1 treatment is to achieve adult height within the individual’s target range. However, SPIGFD can also affect functional capabilities and negatively impact daily activities. For example, individuals with short stature may be unable to drive or apply for certain jobs, such as becoming a flight attendant, where the minimum height requirement is 4 feet 11 inches. Evidence also suggests that IGF-I is important for cell growth and cardiopulmonary function, thereby reducing the risk of cardiac arrest and acute lung injury. Therefore, low circulating levels of IGF-I may have serious health implications for SPIGFD patients beyond height.
Unfortunately, the non-height-related impacts of IGF-I deficiency are poorly understood, and research is limited, leading to the misconception that SPIGFD is primarily a height-related disease. This, in turn, may contribute to the need for comprehensive monitoring of SPIGFD globally and recognition of the tangible impacts of this condition on patients’ health and daily activities. By revealing the non-height-related consequences of IGF-I deficiency, it may be possible to generate broader interest in SPIGFD from industry and policymakers.
Sadly, short-statured children are often raised by their caregivers and teachers, while short-statured adults are frequently stigmatized in social and clinical settings, often perceived as having diminished cognitive abilities. The GH-IGF axis plays a role in normal brain growth and structure, and severe GH deficiency is associated with neurocognitive changes in some patients (possibly related to frequent hypoglycemic episodes). However, normalizing derogatory terms through academic journals and discussions reinforces the inaccurate notion that short-statured individuals are ‘abnormal’ and leads to misunderstandings about the intelligence of SPIGFD patients. It is imperative to overcome misconceptions about short-statured patients to improve care for SPIGFD patients. Therefore, efforts should be made in the medical field and in schools and workplaces to abolish derogatory terms and assumptions about SPIGFD and other short-statured patients.
Burden of Disease
Studies on the quality of life of short-statured children indicate that they have lower self-esteem and attention compared to average-height children. Short-statured children often face bullying and discrimination in school environments due to their height; this may persist into adulthood, potentially affecting their ability to find life partners or employment. Particularly in developing countries, this can significantly impact patients’ ability to earn a living and provide food for themselves and their families. Studies also indicate that short stature affects the psychological and social health of SPIGFD patients. Due to social and economic challenges impacting their daily lives, it is not uncommon for individuals to experience emotional distress and depression. It is crucial that individuals affected by SPIGFD receive the necessary support and counseling throughout childhood, adolescence, and adulthood.
Prior to and during diagnosis, the process can be particularly challenging for patients and their families. Some parents may blame themselves, wondering what they could have done differently. Parents may even worry about their child’s ability to live independently, and concerns about the social-psychological impacts of short stature are key motivating factors for parents seeking medical help. The lack of information and understanding may lead to uncertainty and anxiety for affected families as they seek a clear diagnosis and treatment for their child. As experienced by many patients with rare diseases, prolonged referral processes can be distressing, particularly for young children and their families. Raising awareness of this condition means that patients can receive timely and formal diagnoses, along with adequate genetic counseling, providing comfort and hope for the future.
For SPIGFD patients and their families, initiating treatment can also be a burdensome process, often requiring substantial time and financial costs. For example, if blood glucose control issues are anticipated, UK clinicians often see patients admitted and allocate ~5 days to initiate treatment, providing additional family support as resources allow. In contrast, in the U.S. and other European countries like Poland and Sweden, treatment initiation occurs in an outpatient setting. The benefit of starting treatment in an outpatient setting is that it signals to patients and their families that the condition is manageable without the need for hospitalization. However, the downside of this approach is that patients may need to attend multiple appointments, imposing additional financial and time costs on patients and their families. Additionally, in these children, hypoglycemic events may be less easily detected, although regular monitoring of capillary blood glucose by patients or their caregivers may help track the occurrence of such events.
Once diagnosed, the initiation of treatment itself requires substantial resources and guidance for caregivers in home management, blood glucose monitoring, and injection site reaction management. Given the burden of performing subcutaneous injections twice daily, rhIGF-1 treatment may place stress on family activities, with group members reporting that siblings often receive less parental attention than SPIGFD patients. Given this burden, it is crucial for families to understand the expectations and requirements of treatment before initiating therapy. Healthcare providers should explain to caregivers that maintaining treatment adherence is vital for improving growth in SPIGFD children; one study indicated that over a follow-up of 10.0 years (n=21), rhIGF-1 led to an overall height increase of 13.4cm greater than expected for SPIGFD patients (n=21). Therefore, clearly communicating the requirements and expected long-term benefits is essential to ensure that parents (and subsequently the patients themselves) receive adequate support to undergo treatment.
Future Directions
Based on the results of the multi-stakeholder meeting (Figure 2), it is clear that targeting the right audiences is foundational to improving the diagnosis and treatment of SPIGFD. Specifically, raising awareness among primary healthcare providers should be a priority, as enhancing awareness and understanding of SPIGFD within the healthcare community may provide a framework for improving access to information for those directly affected by SPIGFD as well as the broader society.
Developing updated expert consensus statements on the diagnosis and treatment of SPIGFD can provide much-needed guidance for HCPs. Clear guidance on the most appropriate diagnostic examinations must be provided to enable HCPs to confidently initiate treatment in a timely manner or refer patients to specialists. Similarly, algorithms for the diagnosis and management of IGF-I deficiency will provide useful resources illustrating the potential of various diagnostic tools to characterize SPIGFD and other forms of IGF-I deficiency. There is also a need to collect more data through patient registries to better describe the complexities and treatment responses of global IGF-I deficiency. The drafting of these materials will benefit from the support of international organizations dedicated to advancing research and education related to growth disorders. Combined with well-managed compassionate use programs and the dissemination of accessible information, this will facilitate a better understanding of international IGF-I deficiency and help improve opportunities for diagnosis, access to treatment, and the overall care burden for patients worldwide.
Clearly, there is a need to challenge the current perceptions of SPIGFD. Recognizing the physical, emotional, and social impacts of SPIGFD on patients and their families is a good starting point. Support and counseling should be provided for children with SPIGFD and maintained throughout adolescence, higher education, and adulthood. Additionally, shifting the treatment goals for SPIGFD from solely increasing height to improving overall health will be key to enhancing awareness and opportunities for care for SPIGFD patients.
Conclusion
This article summarizes the outcomes of the multi-stakeholder meeting, highlighting the challenges faced by patients, families, and healthcare providers regarding SPIGFD. As a rare endocrine disease, the diagnostic and treatment pathways are unclear, and awareness of this condition is limited, often resulting in delayed diagnosis (and subsequent treatment). Group members emphasized the need for education and clearer guidance for healthcare providers to facilitate early diagnosis and the initiation of appropriate treatment, with the ultimate goal of improving clinical outcomes and quality of life for SPIGFD patients. Efforts to identify solutions to promote equitable access to appropriate care are particularly important in regions where patients struggle to obtain diagnosis and proper treatment due to weak infrastructure, political turmoil, regional conflicts, and/or economic barriers.
The main recommendations from the multi-stakeholder meeting include developing widely accessible materials to educate and challenge existing perceptions around SPIGFD, shifting the focus from height to overall measurable outcomes of the condition, and providing valuable guidance and support. Additionally, ongoing efforts by clinicians, patient advocacy groups, medical societies, and other key stakeholder groups to raise awareness and understanding of this complex disease globally are essential to address many of the challenges raised in this article and improve overall care for SPIGFD patients.
Chen Kang 2024.06
Endocrine Metabolic Disease @CK Medicine
Endocrine Metabolic Disease Knowledge Framework @CK Medicine
Endocrine Metabolic Disease Tiered Diagnosis and Treatment @CK Medicine
CK Note: Why does this public account emphasize the promotion of guidelines or consensus?