Shengqi Huo 1, Qian Wang 1, Wei Shi 1, Lulu Peng 1, Yue Jiang 1, Mengying Zhu 1, Junyi Guo 1, Dewei Peng 1, Moran Wang 1, Lintong Men 1, Bingyu Huang 1, Jiagao Lv 1, Li Lin 12023 Jan 14;24(2):1667. doi: 10.3390/ijms24021667.
Affiliationsexpand PMID: 36675183 PMCID: PMC9862315 DOI: 10.3390/ijms24021667
Research Background
The acquisition, distribution, and elimination of trace metals such as iron (Fe) and copper (Cu) are crucial for life. Metabolic diseases such as diabetes, obesity, and non-alcoholic fatty liver disease often involve the interaction of multiple potential molecular mechanisms. Multiple lines of evidence suggest that metabolic diseases, including obesity and diabetes, are often accompanied by dysregulation of various metal ions. Among these, copper is central to many important biological processes, including mitochondrial respiration, antioxidant defense, and biosynthesis of biomolecules, and is particularly closely related to the severity and progression of diabetes. Serum copper is significantly elevated in diabetic patients and in STZ-induced diabetic rat models. In vivo and in vitro experiments show that low concentrations of copper ion coronary perfusion can significantly reduce cardiac function, while the divalent copper chelator triethylenetetramine can significantly improve cardiac function in diabetic cardiomyopathy rats. This experimental evidence suggests that copper homeostasis may play an important role in maintaining cardiac function. However, how copper ions affect cardiac function in diabetic cardiomyopathy remains elusive.
Recently, a unique insight into programmed cell death (referred to as copper death) has been proposed in tumor cells and ATP7B – / – mice. This copper-induced cell death is characterized by a reduction in the lipoylation of DLAT and DLST, as well as oligomerization of TCA cycle lipoylated proteins induced by direct binding of copper, while the instability and overall reduction of iron-sulfur (Fe-S) cluster proteins together lead to protein toxic stress and mitochondrial dysfunction. Iron-sulfur cluster protein FDX1 and lipoylation are key regulators of copper death. However, copper death in cardiomyocytes has not been reported, and whether diabetic cardiomyopathy involves copper death remains undetermined.
Copper maintains precise balance in all organisms through mechanisms of uptake, transport, storage, and excretion. Loosely bound copper is restricted to a nearly undetectable low level, with less than one atom per cell. The redox copper (II) bound to plasma protein carriers (copper ceruloplasmin) predominates in the blood. Cu(II) is reduced to Cu(I) by reductases and transported into the cytoplasm by the high-affinity SLC31A1 (CTR1). The incoming Cu(I) binds to metallothioneins (such as GSH) and metal chaperones (such as superoxide dismutase), distributing Cu(I) to different subcellular locations. Copper ion clusters (such as chlorobutanol) are small molecules that can deliver copper into cells, disregarding ion concentration gradients. Therefore, they are useful tools for studying copper toxicity. A series of studies have shown that the mechanism by which copper ion clusters induce cell death involves the accumulation of intracellular copper, rather than the action of the small molecule chaperones themselves.
Advanced glycation end-products (AGEs) are a heterogeneous group of compounds, including more than 20 different products generated through glycation, known as the Maillard reaction. Glucose, fructose, or more reactive dicarbonyls undergo non-enzymatic reactions with nucleophilic groups on proteins, preferentially reacting with the ε-amino group of lysine and N-terminal amines. Intermediate compounds undergo a series of rearrangements and are subsequently converted into stable AGEs. AGEs react with new proteins, perpetuating and propagating oxidative modifications and generating new AGEs crosslinks, which gradually accumulate. Although this reaction is very slow under physiological conditions, under the high glucose conditions of diabetes, the modification of proteins by glucose is significantly accelerated, and is considered one of the main factors in the occurrence of diabetic complications. More importantly, copper-catalyzed oxidation reactions involving Cu 2+ and iron 3+ redox metal ions participate in the formation of AGEs under in vivo conditions; however, these metal ions are sequestered in specific metal transport proteins and other metalloproteins in plasma or cytoplasm. The copper metalloproteins undergo significant conformational changes or even fragmentation after glycation, leading to the release of bound metals, completing a positive feedback loop.AGEs can promote cardiomyocyte death through calcium overload, oxidative stress, or excessive autophagy. However, it remains unclear whether copper death is related to AGEs-induced cardiomyocyte death. Therefore, we investigated the potential impact of copper death on the functional impairment of diabetic cardiomyocytes induced by AGEs.
Research Results
Result 1: Copper Ion Carriers Induce Copper Death in Cardiomyocytes, a Unique Regulated Form of Cell Death
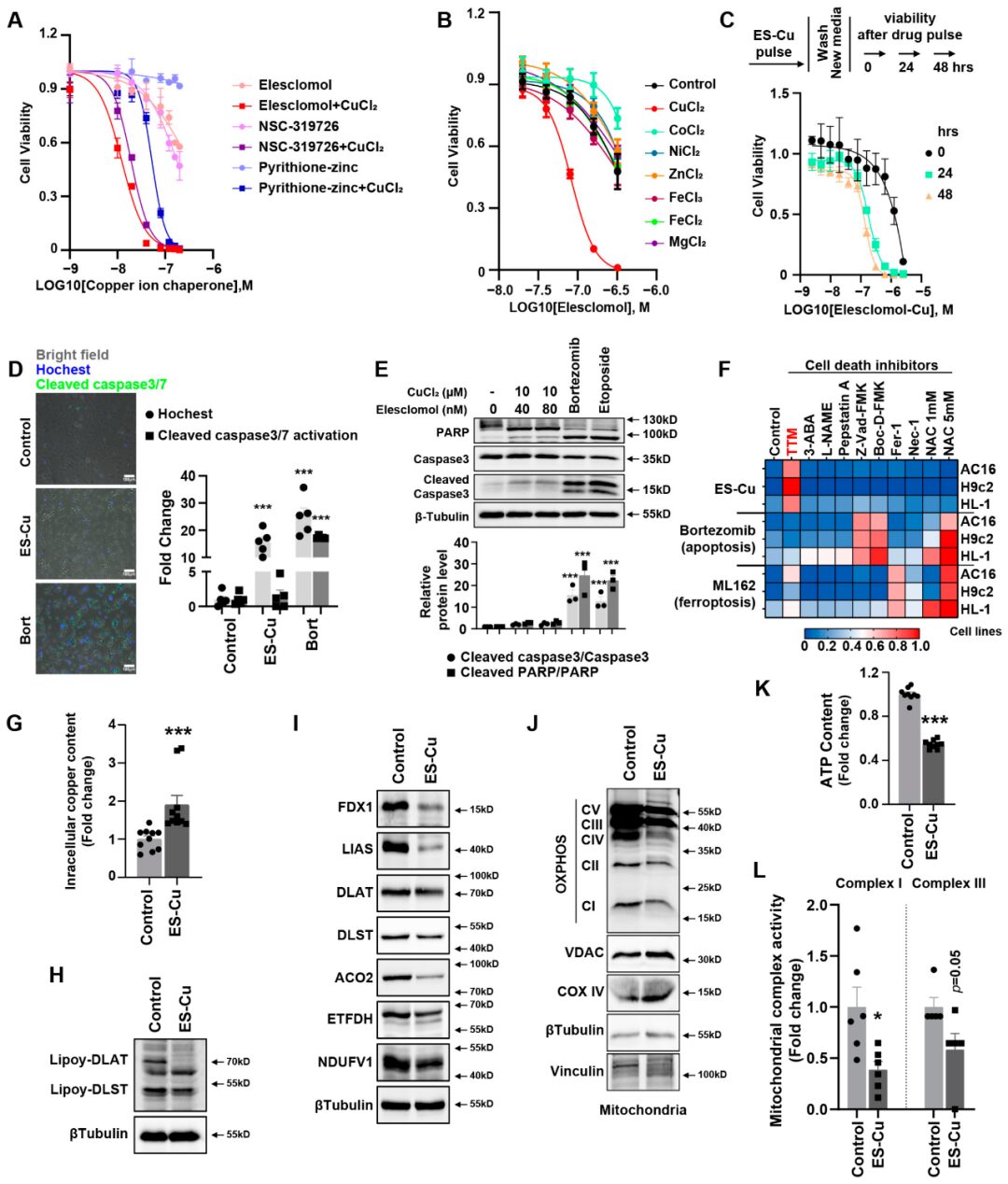
The authors screened the cytotoxic effects of various structurally different copper ion carriers in cardiomyocytes and found that the combination of elesclomol and copper chloride had significant cytotoxic effects on AC16 cardiomyocytes, while copper ion carrier-induced cell death was mild (Figure1A and Supplementary FigureS1). To further confirm that the cytotoxicity of copper ion carriers is selectively dependent on copper, we analyzed the killing potential of the potent copper ion carrier elesclomol in combination with various metal ion chlorides. Only copper exacerbated the cytotoxic effects in cardiomyocytes, while the addition of other metals, including iron, cobalt, zinc, magnesium, and nickel, failed to enhance cell death compared to treatment with elesclomol alone (Figure1B). This indicates that elesclomol has the highest selectivity for copper. Two hours of pulse treatment with elesclomol-copper chloride significantly triggered cell death (Figure1C). At the same time, treating the cells with a two-hour pulse of elesclomol-copper chloride (ES-Cu) followed by washing with fresh media to remove ES-Cu and culturing these cells for another 24 or 48 hours showed that cell viability decreased with continued culture (Figure1C). This result indicates that short-term exposure to copper-containing copper ion carriers leads to irreversible subsequent cytotoxicity.
We used caspase 3/7 positive cells to assess the level of apoptotic cells. Caspase 3/7 positive cells exhibited green fluorescence. The total cell count in bright field images was calculated using ImageJ software. The quantification of caspase 3/7 activity was based on the comparison of the number of green fluorescent positive cells to the total cell count. Bortezomib can target multiple pathways, including p53, nuclear factor κB, phosphoinositide 3-kinase pathway, and ubiquitin/proteasome pathway, to promote apoptosis. (Figure1D) shows that bortezomib induced a 20 to 30-fold increase in green positive apoptotic cells. However, treatment with elesclomol-CuCl2 did not increase this. Elesclomol-copper chloride treatment induced cell death without caspase 3/7 cleavage, which is significantly different from bortezomib-induced cell death. At the same time, after treatment with the apoptosis inducers bortezomib and etoposide, cleaved caspase 3 and cleaved PARP were elevated, but there was no increase after elesclomol-CuCl2 treatment (Figure1E). Similarly, when cardiomyocytes were co-treated with pan-caspase inhibitors (Z-VAD-FMK and Boc-D-FMK), the killing potential of elesclomol-CuCl2 was still maintained (Figure1F), again indicating that copper-induced cell death is distinct from apoptosis. Furthermore, treatment with inhibitors of other known cell death mechanisms, including ferroptosis (ferrostatin-1), necroptosis (necrostatin-1), and oxidative stress (N-acetylcysteine), failed to eliminate copper-induced cardiomyocyte death (Figure1F), indicating that its mechanism is different from known cell death pathways in cardiomyocytes. Tetrathiomolybdate (TTM) copper chelation rescued the cytotoxicity of elesclomol-copper chloride treatment. Elesclomol-copper chloride treatment also significantly induced intracellular copper accumulation in AC16 cells (Figure1G). The results indicate that elesclomol-CuCl2 induced cell death involves the accumulation of intracellular copper. These results suggest that copper-induced cell death is indeed regulated, but in cardiomyocytes, it is non-apoptotic, non-ferroptotic, and non-necroptotic.
This copper ion-induced regulated programmed cell death was previously referred to as cuproptosis in tumors, characterized by a reduction in iron-sulfur (Fe-S) cluster proteins and a decrease in mitochondrial enzyme lipoylation. Next, we detected the hallmark changes of copper death in cardiomyocytes after elesclomol-CuCl2 treatment. The expression of multiple mitochondrial Fe-S cluster proteins was reduced, including FDX1, LIAS, ACO2, ETFDH, and NDUFV1 (Figure1I). So far, only four types of multimeric metabolic enzymes in mammals are known, including pyruvate dehydrogenase complex (PDH), α-ketoglutarate complex (KDH), branched-chain α-keto acid dehydrogenase complex (BCKDH), and glycine cleavage system (GCV), which can undergo lipoylation as a post-translational modification, but these proteins are core to the metabolic field. DLAT and DLST are the E2 components of the PDH and KDH complexes, respectively. The lipoylation of DLAT and DLST is another hallmark of copper death. We also found that after elesclomol-copper treatment in cardiomyocytes, the lipoylation of DLAT and DLST was reduced (Figure1H). Copper also reduced the expression of mitochondrial respiratory chain complexes (Figure1J). Mitochondrial function, including intracellular ATP content (Figure1K) and the activity of mitochondrial complexes I and III (Figure1L), decreased after incubation with elesclomol-CuCl2. These results indicate that mitochondrial function plays an important role in copper-induced cardiomyocyte death.
Result 2: AGEs Induce Copper Death in Cardiomyocytes
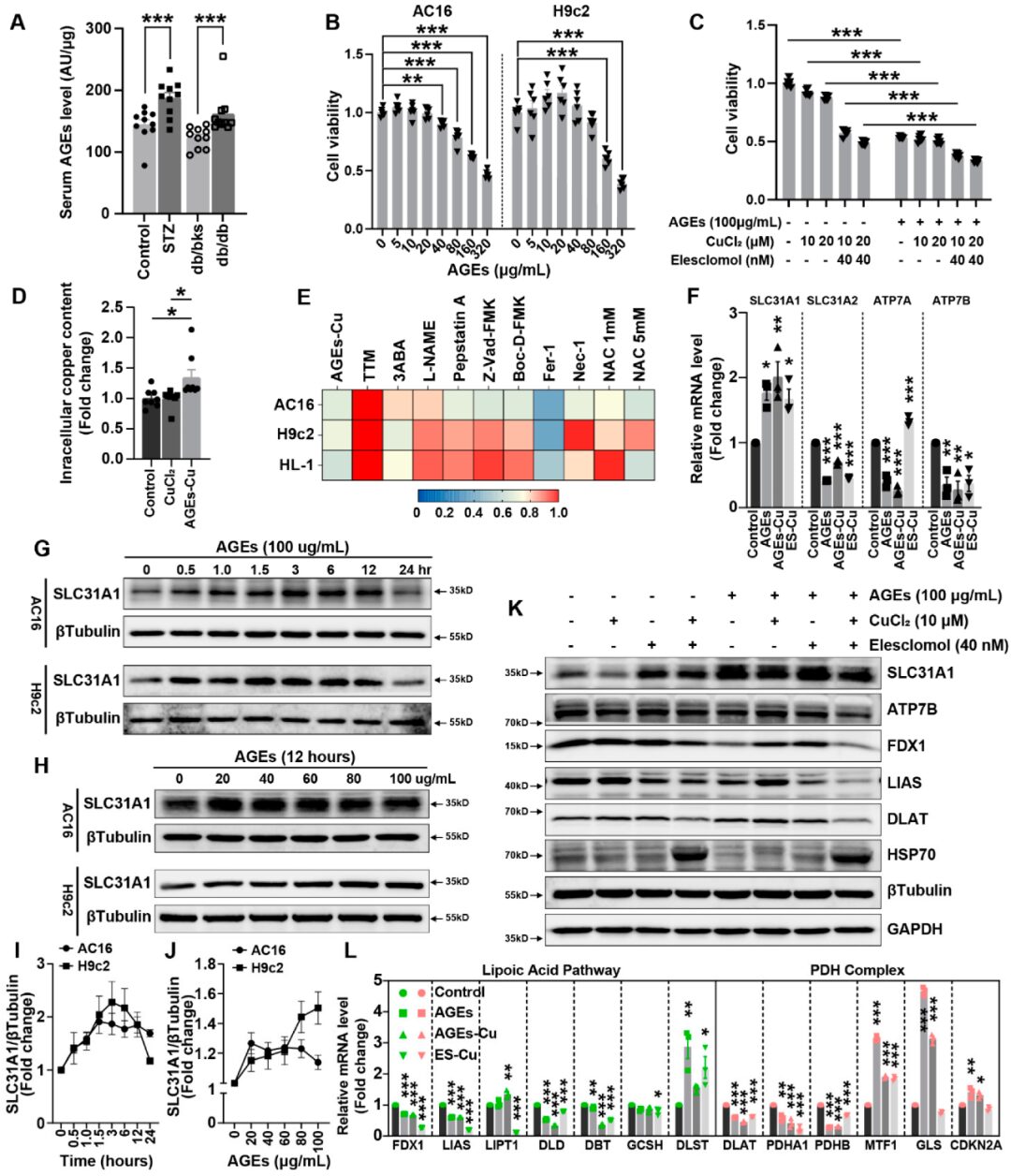
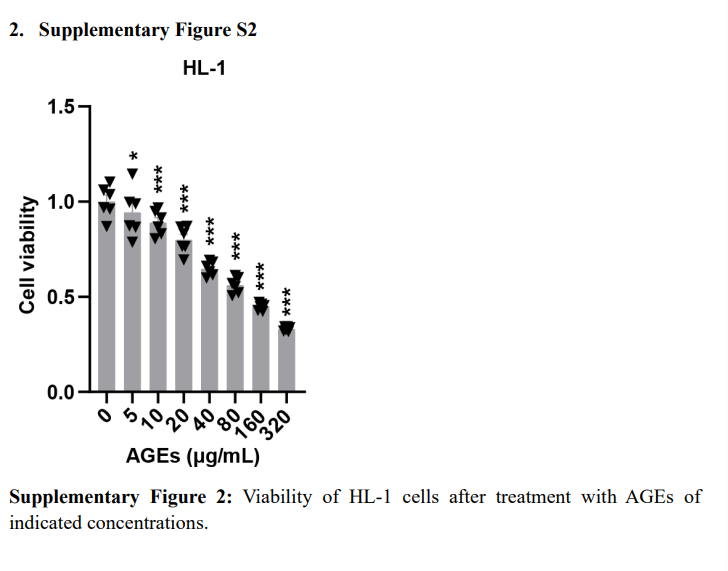
Serum AGEs levels in STZ-induced and db/db diabetic mice were significantly elevated (Figure2A). AGEs significantly induced cell death in AC16, H9c2, and HL-1 cardiomyocytes in a concentration-dependent manner (Figure2B and Supplementary FigureS2). AGEs, CuCl2, or elesclomol-CuCl2 alone promoted cell death in AC16 cells, while the combination of AGEs with CuCl2 or elesclomol-CuCl2 exacerbated cell death in AC16 cardiomyocytes (Figure2C). AGEs treatment increased copper accumulation in cardiomyocytes, indicating that copper is related to AGEs-induced cell death (Figure2D). Using various cell death mechanism inhibitors also suggested that the alleviation of AGE-CuCl2-TTM induced cardiomyocyte death was more pronounced than with other inhibitors (Figure2E).
Secondly, AGEs treatment, with or without CuCl treatment, increased the mRNA expression of the copper input gene SLC31A1, while the expression of copper output genes ATP7A and ATP7B decreased (Figure2). Similarly, the protein expression of SLC31A1 increased with prolonged incubation time of AGEs or increased concentrations of AGEs in AC16 and H9c2 cells (Figure2G,H). These indicate that AGEs increase the protein expression of SLC31A1 in a concentration-dependent and time-dependent manner in cardiomyocytes (Figure2I,J). Fe-S cluster proteins FDX1 and LIAS are markers of copper death, which were reduced after elesclomol-CuCl2 treatment. Elesclomol-CuCl2 treatment also significantly induced the expression of SLC31A1 and HSP70 in cardiomyocytes (Figure2K). Notably, the combination of AGEs significantly promoted the changes in the expression of copper death-related proteins compared to treatment with elesclomol-CuCl2 alone (Figure2K). The lipoic acid pathway and PDH complex are both important pathways involved in copper death. RT-PCR analysis indicated that the changes induced by AGEs-CuCl2 treatment were similar to those induced by elesclomol-CuCl2 treatment (Figure2L). At the same time, the changes in the PDH complex pathway induced by AGEs-CuCl2 treatment were also similar to those induced by elesclomol-CuCl2 treatment. These data indicate that AGEs can induce copper death in cultured cardiomyocytes.
Result 3: AGEs Regulate SLC31A1 Expression in AC16 Cardiomyocytes via ATF31/SPI1
Integrating ALGGEN PROMO, ChIP Atlas, Cistrome DB, GeneCards, GTRD, hTFtarget, and UCSC JASPAR databases, potential transcription factors for SLC31A1 were predicted. The inverted Venn diagram from seven databases revealed a total of seven potential transcription factors: ATF2, ATF3, SPI1, ELF1, IRF1, RELA, and HNF4A (Figure3A). Next, we utilized the hTFtarget database to predict potential co-transcription factors of the cuproposis gene set, including FDX1, LIAS, LIPT1, DLD, DLAT, PDHA1, PDHB, MTF1, GLS, and CDKN2A. The intersection of the two predicted TF sets indicates that ATF3 and SPI1 may play a role in the transcriptional regulation of SLC31A1 and the copper gene set (Figure3B).
Next, we validated the potential roles of SPI1 and ATF3 in AC16 cardiomyocytes. After treatment with AGEs, with or without CuCl2, the expression of SPI1 and ATF3 mRNA increased by approximately 20 or 30 times in AC16 cardiomyocytes (Figure3C). The changes in other transcription factors ATF2, ELF1, IRF1, RELA, and HNF4A were not as pronounced as those of ATF3 and SPI1 (Figure3C). In AC16 cells, the expression of both transcription factors ATF3 and SPI1 also increased in a time- and concentration-dependent manner with prolonged incubation time or increased concentrations of AGEs (Figure3D,E). After treatment with AGEs, the expression of ATF3 and SPI1 proteins showed time and concentration-dependent increases (Figure3F,G). Meanwhile, the combination of AGEs significantly promoted the expression of SPI1 and ATF3 proteins compared to treatment with elesclomol-CuCl2 alone (Figure3H). However, the increase in protein expression of RELA and IRF1 was not significant compared to that of ATF1 and SPI3 (Figure3H). Overexpression of ATF3 and/or SPI1 significantly increased the mRNA and protein expression of SLC31A1 in cardiomyocytes (Figure3I,J).
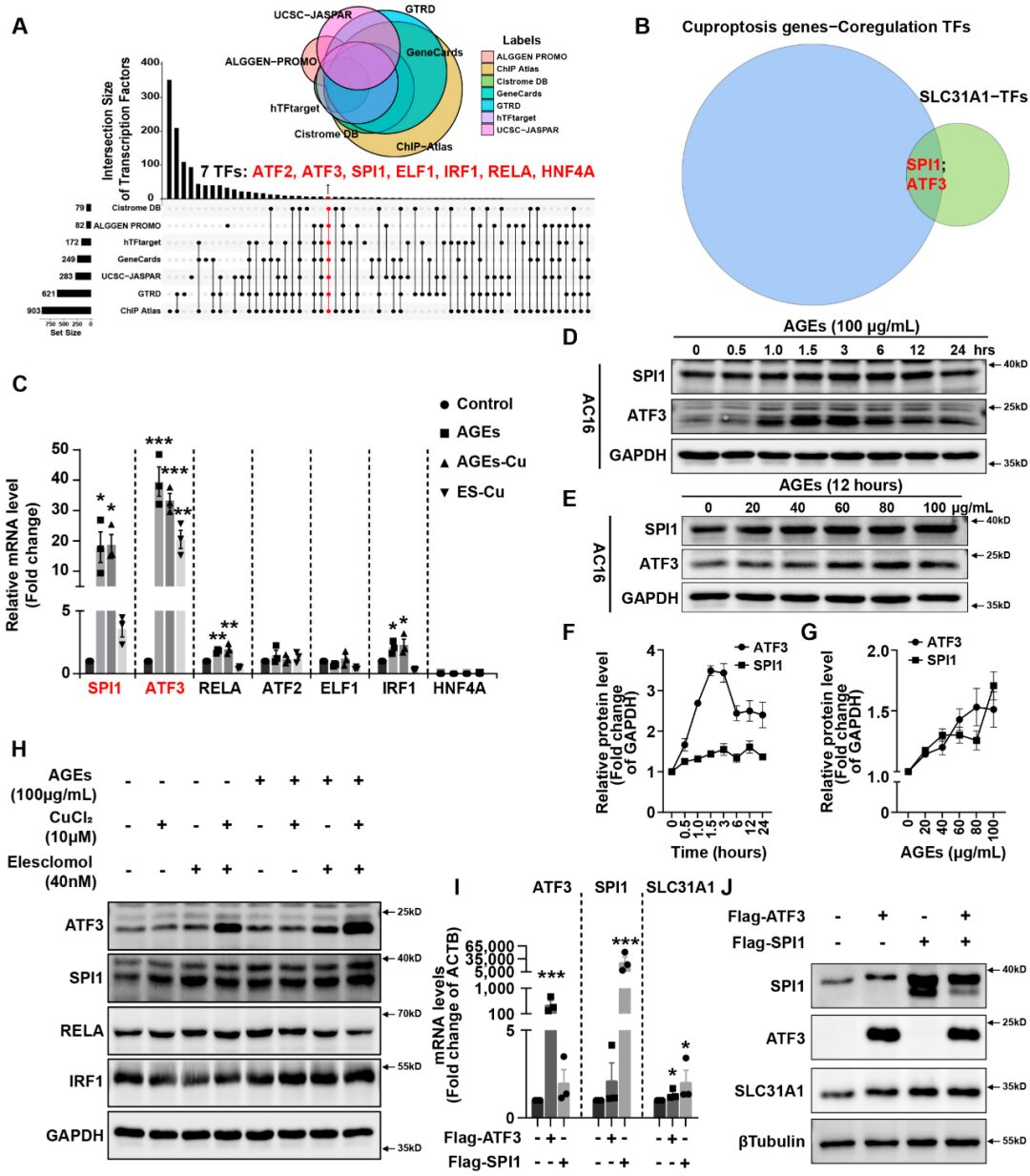
Result 4: AGEs/SPI1/ATF3/SLC31A1 Signaling Downregulates Mitochondrial Iron-Sulfur Cluster Protein Expression and Inhibits Mitochondrial Function
Previous studies have reported that the decrease in iron-sulfur (Fe-S) proteins is a key outcome of elesclomol-CuCl2 treatment. The biogenesis of cellular iron-sulfur (Fe/S) proteins is a fundamental and minimal function of mitochondria. This process is catalyzed by the bacterial-derived iron-sulfur cluster assembly (ISC) mechanism and has been divided into three main steps: de novo synthesis of (2Fe-2S) clusters on scaffold proteins; cluster transport mediated by Hsp70 chaperones and insertion into target apoproteins; and the catalytic conversion of (2Fe-2S) clusters into (4Fe-4S) clusters, which are then inserted into recipient apoproteins. The ISC components in the first two steps of this core ISC mechanism are also essential for the biogenesis of many required cytoplasmic and nuclear Fe/S proteins, which explains the essence of mitochondria. The third step, also known as the late ISC mechanism, dominates in mitochondrial Fe/S proteins. Therefore, we examined the changes in Fe-S clusters after AGEs-CuCl2 treatment to investigate whether Fe-S clusters are involved in diabetic myocardial injury. RT-PCR was used to detect the mRNA changes of 20 mitochondrial Fe-S cluster genes involved in mitochondrial function after supplementation with AGEs and AGEs-CuCl2. The mitochondrial respiratory chain proteins NDUFS7, NDUFS8, and NDUFV1 as well as ACO2 and ETFDH, which are involved in the tricarboxylic acid cycle and β-oxidation, were significantly downregulated (Figure4A). We also found that core ISC machinery proteins NFS1, ISD11, and FDXR, late ISC machinery proteins NFU1, BOLA3, and IND1, as well as transport proteins ABCB7 were significantly reduced after AGEs and AGEs-CuCl2 treatment (Figure4B).
LIAS is a regulator of lipoic acid metabolism and lipoylation; mitochondrial proteins DLAT and DLST lipoylation and oligomerization affect mitochondrial function. Previous studies reported that copper directly binds and promotes the oligomerization of lipoylated DLAT, thereby inhibiting mitochondrial function. Interestingly, we also found that AGEs or AGEs-CuCl2 had significant effects on lipoic acid metabolism in cardiomyocytes. The expression of lipoylation of DLAT, DLST, LIAS, and DLAT and DLST was reduced (Figure4C). At the same time, the oligomerization of DLAT and DLST was induced by AGEs or AGEs-Cu (Figure4C). AGEs and AGEs-CuCl2 also similarly reduced the expression of Fe-S cluster proteins NDUFS8, NDUFV1, ETFDH, ACO2, NFU1, NFS1, and ABCB7 (Figure4D). Overexpression of ATF3 and SPI1 also played a similar role in reducing the expression of Fe-S cluster proteins FDX1, ACO2, and DLST in cardiomyocytes (Figure4E).
The protein blot analysis of mitochondrial oxidative respiratory chain also confirmed that AGEs inhibit mitochondrial respiration (Figure4F). At the same time, AGEs and AGEs-CuCl2 reduced the ATP content in cardiomyocytes (Figure4G) as well as the activity of mitochondrial complexes I and III (Figure4H,I). These results indicate that AGEs inhibit mitochondrial function, which is associated with the decrease in Fe-S clusters.
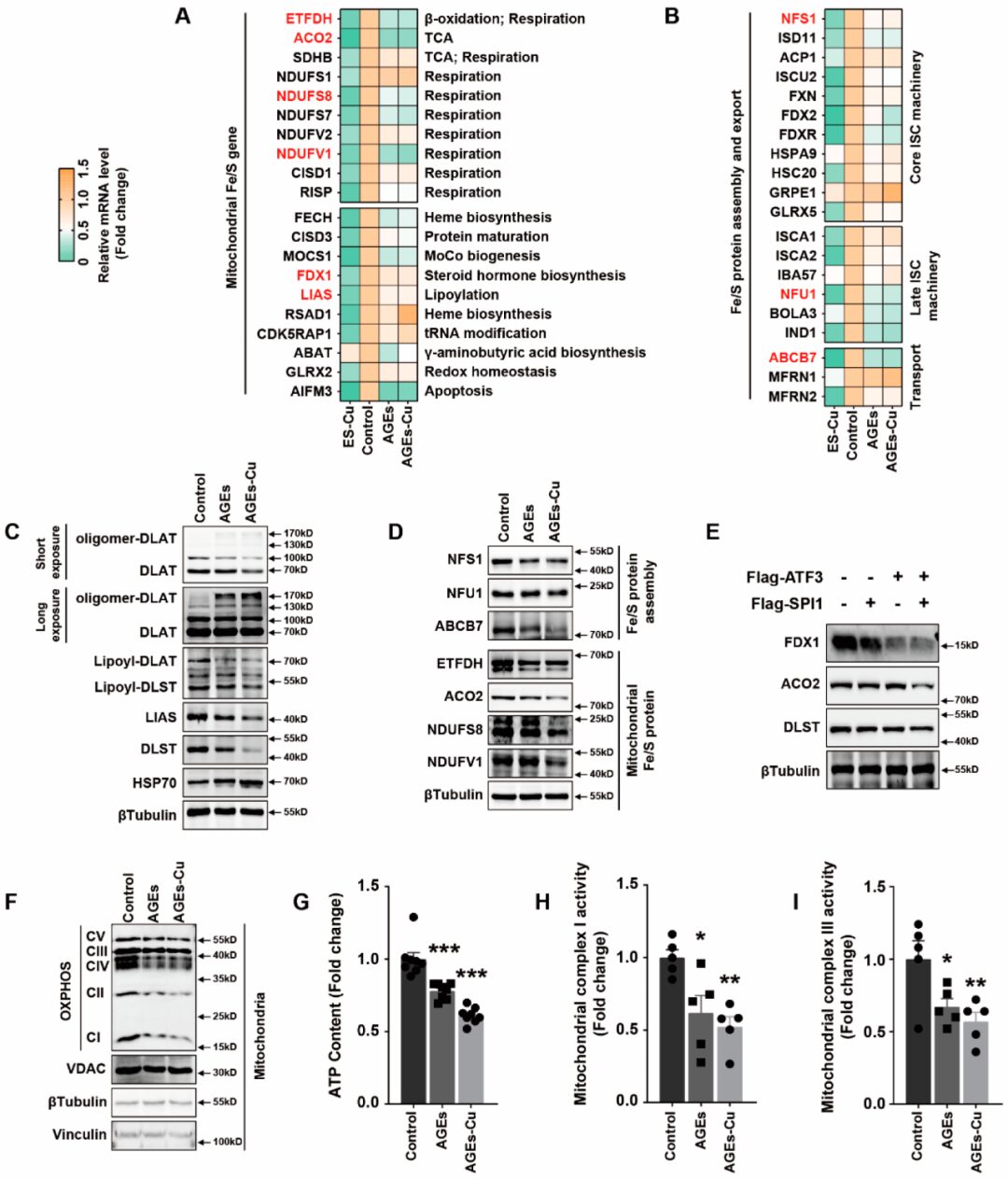
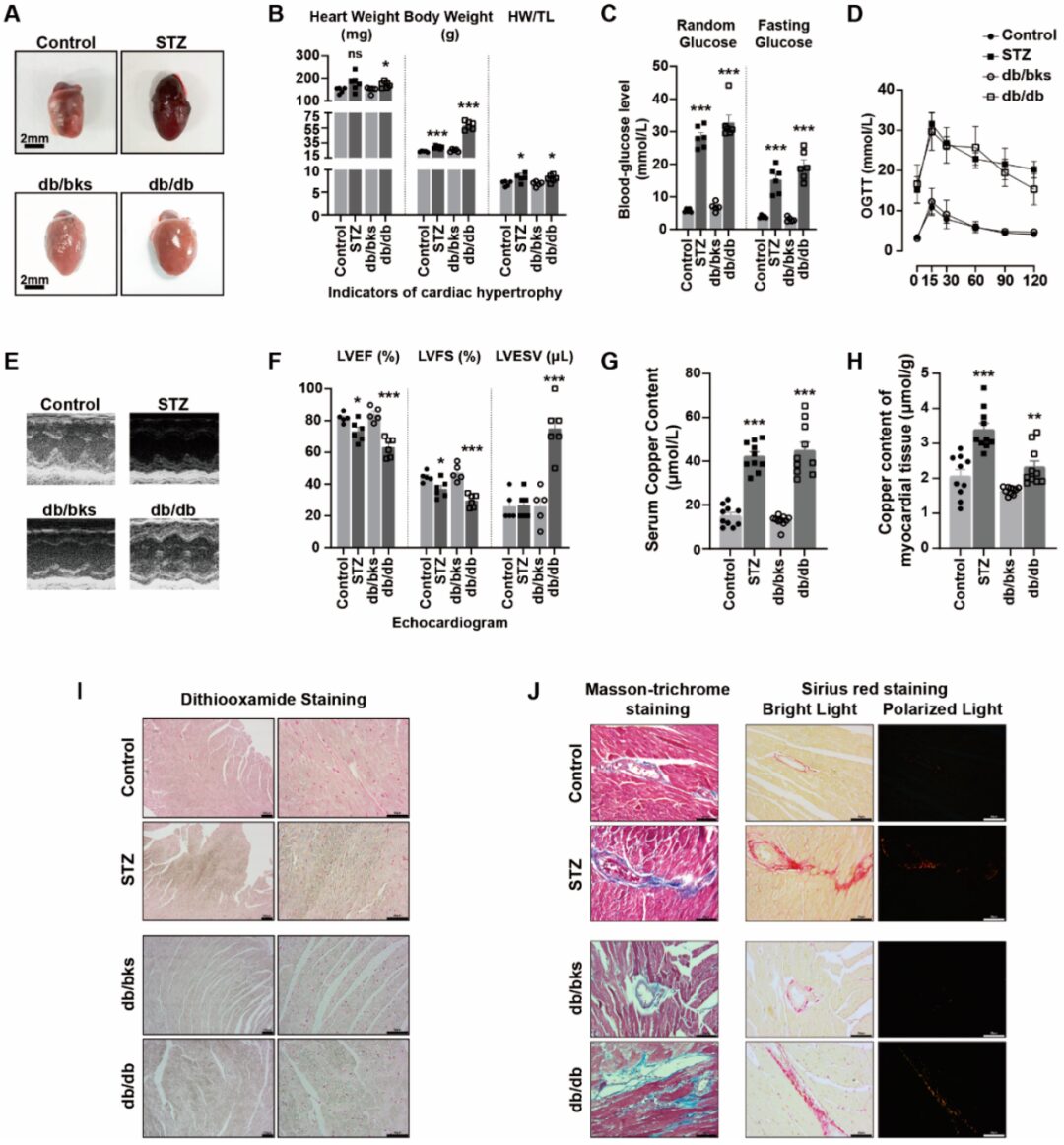
Result 5: Elevated Copper Levels in Peripheral Blood Circulation and Myocardial Tissue of Diabetic Mice Exacerbate Myocardial Injury
STZ-induced diabetes and db/db spontaneous diabetic mice exhibited cardiac enlargement (Figure5 A), heart weight (HW), body weight (BW), and heart weight/tibial length (HW / TL) ratios greater than those of control or db/bks mice (Figure5B). Random blood glucose, fasting blood glucose, and oral glucose tolerance test (OGTT ) experiments indicated abnormal glucose levels and glucose tolerance in diabetic mice (Figure5C,D). Chest echocardiography also confirmed that the left ventricular ejection fraction (LVEF) and left ventricular fractional shortening (LVFS) were reduced, while the left ventricular end-systolic volume (LVESV) was significantly increased (Figure5E,F), indicating impaired cardiac function in diabetic mice. Masson’s trichrome staining and Sirius red staining showed that the degree of myocardial fibrosis was significantly increased in diabetic mice (Figure5J).
The copper ion levels in the serum and myocardial tissue of diabetic mice were significantly higher than those in the control group (Figure5G,H). Disulfide oxalate staining was used to identify the accumulated copper salts in tissues. We found that diabetes exacerbated the accumulation of copper salts in myocardial tissues (Figure5I). These results suggest that copper may be involved in the process of diabetic myocardial injury.
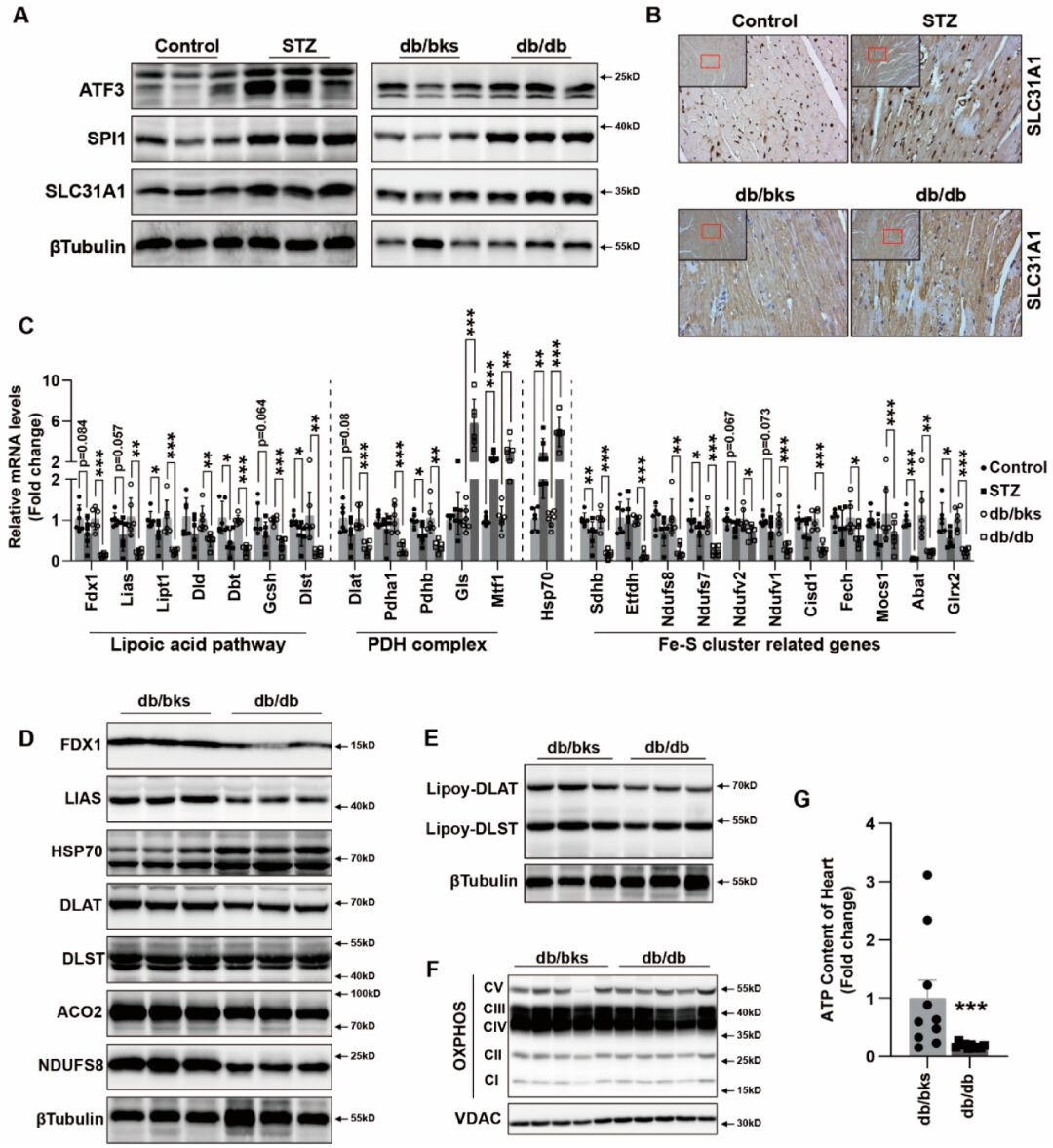
Result 6: ATF3/SPI1/SLC31A1 Copper Death Signaling Involves Myocardial Injury in Diabetic Mice
ATF3, SPI1, and SLC31A1 were increased in diabetic myocardial tissues, indicating that the ATF3 / SPI1 / SLC31A1 signaling pathway was activated (Figure6A). We performed immunohistochemical staining for SLC31A1 and found increased expression in myocardial tissues (Figure6B). Changes in the levels of lipoic acid pathway, PDH complex, and Fe-S cluster gene mRNA in STZ and db/db diabetic mice were consistent with those in vitro in cardiomyocyte cuproposis (Figure6C). We also observed the loss of Fe-S cluster proteins FDX1, LIAS, NDUFS8, and ACO2 in the hearts of db/db mice, along with an increase in HSP70 abundance (Figure6D). Meanwhile, the reduction of DLAT and DLST expression as well as the decrease in lipoylation of DLAT and DLST were also significant in the hearts of db/db mice (Figure6D,E). Compared to db/bks mice, the protein expression of mitochondrial oxidative respiratory chain complexes was also decreased in the hearts of db/db mice (Figure6F). At the same time, the ATP content in the heart tissues of db/db mice was reduced (Figure6G). These findings in the copper toxicity diabetic mouse model suggest that copper overload leads to cellular effects similar to those induced by AGEs combined with copper ion carriers. Taken together, our data support that excess copper promotes the instability of Fe-S cluster proteins, leading to mitochondrial dysfunction and ultimately cell death.
Copper (Cu) overload-induced cuproptosis has not been studied in diabetic cardiomyopathy (DCM). Advanced glycation end-products (AGEs) induced by sustained hyperglycemia play a crucial role in cardiac toxicity. To elucidate whether AGEs-induced cardiac toxicity is related to CUPROPOSIS, the authors analyzed the toxicity of AGEs and copper in AC16 cardiomyocytes and STZ-induced or db/db diabetic mouse models. The results indicated that the copper ion carrier elesclomol induces copper death in cardiomyocytes. It was rescued only by the copper chelator tetrathiomolybdate, but not by other cell death inhibitors. Interestingly, AGEs triggered the accumulation of copper in cardiomyocytes and exhibited features of copper death, including the loss of Fe-S cluster proteins (FDX1, LIAS) and exacerbated cardiomyocyte death when incubated with CuCl2. Furthermore, AGEs increased the loss of intracellular S, NDUFS8, and ACO2 as well as a decrease in lipoylation of DLAT and DLST. These effects were accompanied by reduced mitochondrial oxidative respiration, including downregulation of mitochondrial respiratory chain complexes, reduced ATP production, and suppressed activity of mitochondrial complexes I and III. Additionally, AGEs promoted the upregulation of SLC31A1. We predicted that ATF3 and/or SPI1 might be transcription factors for SLC31A1 through online databases, and validated this through overexpression of ATF3 and/or SPI1. In diabetic mice, increased copper and AGEs were observed in blood and heart, accompanied by heart failure. Changes in the protein and mRNA profiles in diabetic heart were consistent with copper death. Our findings first indicate that excessive AGEs and copper in diabetes upregulate the ATF3 / SPI1 / SLC31A1 signaling pathway, disrupting copper homeostasis and promoting copper death. Overall, this novel mechanism may serve as an alternative potential therapeutic target for DCM.
Research Conclusion
1. The copper ion carrier elesclomol induces copper death in cardiomyocytes.
2. AGEs trigger the accumulation of copper in cardiomyocytes, exhibiting features of copper death.
3. Excessive AGEs and copper in diabetes upregulate the SLC31A1 signaling pathway, disrupting copper homeostasis and promoting copper death.
Proofread by: Dong Chao