Hello everyone, today I will share an article published in BMC Bioinformatics. The corresponding authors of the article are Associate Professor Raquel C. de Melo-Minardi, Dr. Ronaldo A. P. Nagem, and Dr. Diego Mariano from Universidade Federal de Minas Gerais, Brazil.

The study of the effects of protein mutations may help to better alter the properties of proteins, such as enzymes that require higher thermal stability and conformational stability for industrial applications in biotechnology. However, experimentally detecting mutations is a complex, time-consuming, and costly task. The authors of this article developed Proteus, a web server that can provide mutation suggestions for residue pairs observed in known structures to enhance the stability of target proteins.
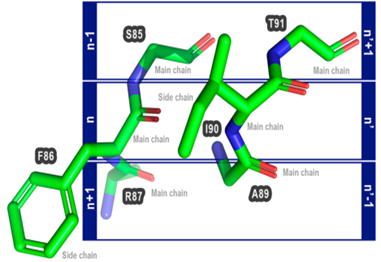
Proteus is based on the assumption that changing a pair of non-interacting amino acid residues to a pair of interacting amino acids can improve the stability of the protein. Proteus can be accessed using a web tool.Proteus takes the 3D structure of a protein as input and then selects amino acids that are close to each other (not in direct contact, <10Å, with chain numbers or specified residues) and their four neighboring residues.By comparing each selected triplet pair in the target protein with triplet pairs in the Proteus database (ProteusDB), potential mutation pairs that could be introduced into the target protein to enhance its stability without significant conformational changes can be searched.Each triplet pair in ProteusDB is formed by the backbone atoms of two interacting residues (n and n’) and their respective neighboring residues, with the coordinates of these residues collected from the PDB database.To reduce redundancy in the triplet pairs in ProteusDB, the authors clustered them using Gromacs’s gmx cluster, ultimately collecting 175,267 triplet pairs.
The PSE in Proteus is used to discover potential beneficial mutation search methods, utilizing Biopython for structural comparisons between target and ProteusDB triplet pairs. If the RMSD is less than 0.5Å, PSE considers that the two residues can be exchanged simultaneously without causing undesirable backbone conformational changes. Comparing with all triplet pairs in ProteusDB would consume a lot of computational resources, so the authors introduced a filtering step using the SSV (structural Signature Variation) method before structural comparisons. SSV is a graph-based three-dimensional structure comparison method, and using SSV as a filter can reduce the number of structural comparisons. The results obtained from the comparisons will also be predicted for their ΔΔG values using MAESTRO software.
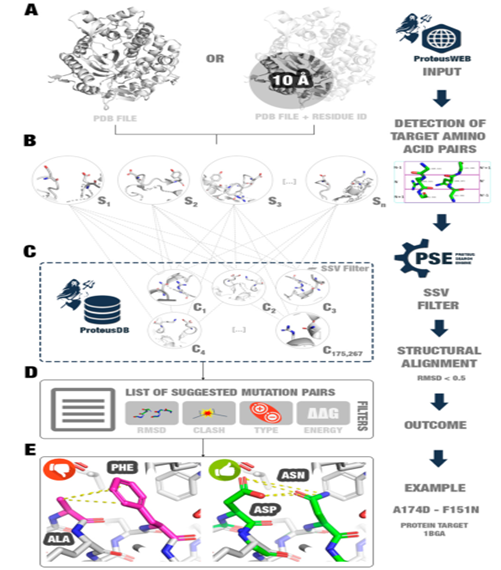
Next, the authors attempted to use Proteus to suggest mutations for several wild-type proteins, analyzing various interactions of the contact residues of the mutants using the Arpeggio script, and found that the number of contact residues in most mutants increased. Among them, the suggested four single-point mutations for the bacteriophage T4 lysozyme structure (PDB ID: 2LZM) received experimental support from the ProTherm database (which has limited related data).
In summary, the authors proposed a new computational method that introduces new side-chain interactions in proteins, providing suggested mutation pairs for the target three-dimensional structure, which can offer new ideas for the rational design of engineered proteins.
Authors: Gu ZH
Original link:
https://bmcbioinformatics.biomedcentral.com/track/pdf/10.1186/s12859-020-03575-6
Original citation: DOI: 10.1186/s12859-020-03575-6
