Good morning, everyone. Today we continue our discussion on “Essential Linux Skills for Bioinformatics Analysis“
In the world of bioinformatics analysis, we deal with various mysterious data files every day: FASTQ files hide the secrets of DNA sequences, GFF annotation files record the location information of genes, and VCF files store the results of variant detection. These files are often massive— a whole genome sequencing FASTQ file can be dozens of GB, while the human genome annotation file can be several hundred MB.
If you try to open these giant files with Notepad or a text editor, either the system will freeze, or the software will crash. So, how can we elegantly view the contents of these “big guys” in a Linux system?
Today we will learn several powerful commands in Linux for viewing file contents. They act like magnifying glasses and telescopes for exploring data treasures, allowing you to quickly and efficiently check the contents of files of any size without causing the system to get bogged down.
Why do we need specialized file viewing commands?
Before diving into specific commands, let’s first understand why we need these specialized tools:
Memory Limitations: Loading large data files entirely into memory consumes a lot of system resources. Efficiency Needs: We usually only need to view part of the file’s content, not the whole thing. Real-time Monitoring: In some cases, we need to monitor real-time changes in files, such as log files. Quick Preview: We need to quickly understand the file format and content structure before processing data.
cat Command: Perfect for Small Files
Basic Usage
cat filename.txtcat sample.fastacat analysis.logCommand Explanation:<span>cat</span> (concatenate) command displays the entire content of a file at once. It is simple and direct, suitable for viewing smaller files.
Useful Parameters
cat -n sequences.fasta # Display line numberscat -A config.txt # Show all control characterscat -s large_file.txt # Compress consecutive blank linesBioinformatics Application Scenarios
# View small configuration filescat config.yaml
# Quickly view sequence informationcat sample.fasta
# View analysis scriptscat run_pipeline.sh
# Concatenate multiple small filescat sample_*.txt > combined.txt**Note:** For large files (over a few hundred KB), it is not recommended to use the <span>cat</span> command, as it outputs all content at once, which may cause the terminal to scroll too fast to read.
less and more: Intelligent Browsers for Large Files
less Command: A Modern File Browser
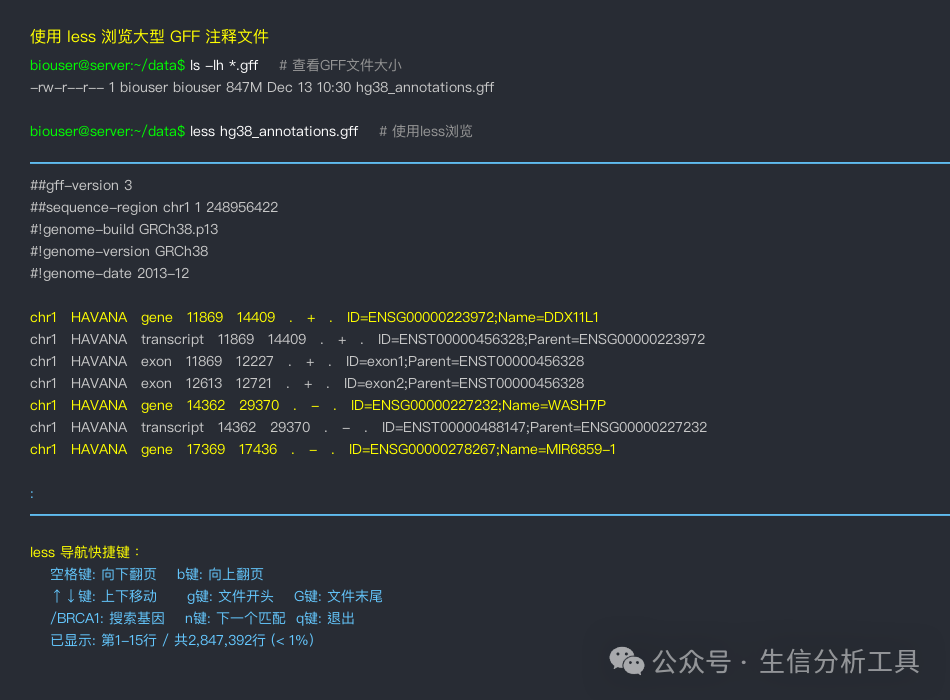
less genome_annotation.gffless large_variants.vcfless /var/log/analysis.logCommand Explanation:<span>less</span> is an interactive file viewer, particularly suitable for browsing large files. It only loads part of the file into memory, allowing it to handle files of any size.
Navigation Shortcuts in less:
- Spacebar: Scroll down a page
- b key: Scroll up a page
- ↑↓ Arrow keys: Move up and down one line
- g key: Jump to the beginning of the file
- G key: Jump to the end of the file
- /pattern: Search down for specific content
- ?pattern: Search up for specific content
- n key: Jump to the next search result
- q key: Exit less
Bioinformatics Practical Tips
# Browse large GFF annotation files, search for specific genesless genome.gff
# In less, type: /BRCA1 (search for the BRCA1 gene)
# View VCF files, jump to the end to see the latest variantsless variants.vcf
# Press G to jump to the end of the file
# Real-time monitor analysis logsless +F analysis.log
# The +F parameter is similar to tail -f, allowing real-time display of new content in the filemore Command: Traditional but Effective
more filename.txtCommand Explanation:<span>more</span> is the predecessor of <span>less</span>, with relatively simple functionality. In systems where <span>less</span> is not available, <span>more</span> remains a reliable choice.
head Command: Peeking at the Secrets of the File’s Start

Basic Usage
head filename.txt # Display the first 10 lines (default)head -n 20 sample.fastq # Display the first 20 lineshead -c 1024 binary_file # Display the first 1024 charactersBioinformatics Application Examples
# Quickly check FASTQ file formathead -n 4 sample_R1.fastq.gz | gunzip# Output:# @SRR123456.1 1 length=150# ATCGATCGATCGATCGATCGATCGATCGATCGATCGATCG...# +# IIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIII...
# View sequence titles in FASTA fileshead -n 20 genome.fasta | grep ">"
# Check header information in VCF fileshead -n 50 variants.vcf | grep "^#"
# View column titles in CSV fileshead -n 1 expression_matrix.csv
# Batch check the beginnings of multiple fileshead -n 5 sample_*.fastqtail Command: Focusing on the Dynamics at the End of Files
Basic Usage
tail filename.txt # Display the last 10 lines (default)tail -n 20 analysis.log # Display the last 20 linestail -c 500 output.txt # Display the last 500 charactersReal-time Monitoring: tail -f
tail -f analysis.log # Real-time display of new content in the filetail -F rotating.log # Handle log rotation situationsCommand Explanation:<span>tail -f</span> is a powerful tool for monitoring real-time changes in files, especially suitable for:
- Monitoring the running logs of analysis programs
- Observing the progress of data processing
- Debugging long-running scripts
Bioinformatics Monitoring Examples
# Real-time monitor BWA alignment progressbwa mem reference.fa sample.fastq > alignment.sam && tail -f bwa.log
# Monitor GATK variant detection outputgatk HaplotypeCaller -I sample.bam -O variants.vcf > gatk.log 2>&1 && tail -f gatk.log
# View the last few records of analysis results tail -n 100 final_results.txt
# Monitor multiple log files tail -f sample_*.logCombining File Viewing Commands
The Power of Piping
# View contents of compressed fileszcat large_file.txt.gz | head -n 20
# Count lines while viewing the beginningwc -l genome.fasta && head -n 10 genome.fasta
# View context of specific content in a filegrep -n "ERROR" analysis.log | head -n 5less +/ERROR analysis.log
# Compare the beginnings of two fileshead -n 20 file1.txt > temp1head -n 20 file2.txt > temp2diff temp1 temp2Practical Bioinformatics Analysis Scenarios
# Check FASTQ file quality encoding formathead -n 1000 sample.fastq | awk 'NR%4==0' | head -n 5
# View gene type distribution in GFF filesless genome.gff | awk '$3=="gene"' | head -n 20
# Monitor progress of multi-sample analysis tail -f samples_*.log | grep "COMPLETED"
# Check alignment result statistics tail -n 50 alignment_stats.txt | grep "mapping rate"Best Practices for Handling Different Bioinformatics File Formats
FASTQ File Viewing Strategies
# Check file integrity (every 4 lines is a sequence unit)head -n 12 sample.fastq # View the first 3 sequences
tail -n 4 sample.fastq # View the last sequence
# Quickly count the number of sequenceswc -l sample.fastq | awk '{print $1/4}'
# Check sequence lengthhead -n 2 sample.fastq | tail -n 1 | wc -cFASTA File Processing Tips
# View sequence titlesgrep ">" genome.fasta | head -n 10
# Count the number of sequencesgrep -c ">" genome.fasta
# View the beginning of sequences for specific chromosomesless genome.fasta# In less, search: />chr1Key Points for Viewing VCF Files
# View header informationhead -n 100 variants.vcf | grep "^#"
# View actual variant data grep -v "^#" variants.vcf | head -n 10
# Check file integritytail -n 1 variants.vcfPerformance Optimization Tips for File Viewing
Handling Compressed Files
# Directly view contents of compressed fileszcat file.txt.gz | head -n 20
zless file.txt.gz
gzip -dc file.txt.gz | tail -n 50Strategies for Handling Large Files
# For particularly large files, use sampling to viewhead -n 1000 huge_file.txt > sample_head.txt
tail -n 1000 huge_file.txt > sample_tail.txt
# Use split to divide large files and then view themsplit -l 10000 large_file.txt chunk_head chunk_aaReal-time Monitoring and Debugging Tips
Simultaneous Monitoring of Multiple Files
# Use multitail tool (requires installation)multitail analysis1.log analysis2.log analysis3.log
# Or use the traditional methodtail -f *.logAdvanced Usage for Log Analysis
# Monitor error messages tail -f analysis.log | grep --color=always "ERROR\|WARNING"
# Monitor progress percentage tail -f progress.log | grep --color=always "Progress: [0-9]*%"
# Real-time count of specific events tail -f access.log | grep "download" | wc -lConclusion
Today we mastered the core tools for viewing file contents in Linux:
- cat: Complete display suitable for small files
- less/more: Interactive browsers for large files, supporting search and navigation
- head: View the beginning of files, quickly understand format and structure
- tail: Focus on the end of files, especially the real-time monitoring function of
<span>tail -f</span>
Key Points:
- Select the appropriate viewing command based on file size
- Master the navigation and search shortcuts in
<span>less</span> - Make good use of
<span>tail -f</span>to monitor long-running analysis programs - Combine piping operations to achieve more complex file content processing
These file viewing commands are fundamental tools in bioinformatics analysis, whether checking data quality, monitoring analysis progress, or debugging program errors. Mastering these commands will allow you to quickly discover key information from vast data files like an experienced detective.
In the next article, we will learn about the first magical tool for text processing—<span>grep</span> command, which can help you accurately search and filter the information you need from massive text.
Practice Suggestions: Find some different formats of bioinformatics files (FASTA, FASTQ, GFF, etc.) and practice viewing them with the commands learned today. Be sure to try the search function in
<span>less</span>and the real-time monitoring of<span>tail -f</span>; these skills are very practical in real work!