“Cancers (Basel)“ published online on July 27, 2022, by Giuseppe Lombardi, Pietro Luigi Poliani, Renzo Manara, and others from Italy, France, Greece, and Switzerland, titled “Diagnosis and Treatment of Pineal Region Tumors in Adults: A EURACAN Overview” (doi: 10.3390/cancers14153646).

Pineal region tumors are rare intracranial tumors, accounting for less than 1% of all adult intracranial tumor lesions. These lesions are a group of histologically heterogeneous tumors. Among these tumors, pineal parenchymal tumors and germ cell tumors (GCTs) are the most common types of lesions.According to the new WHO classification in 2021, pineal parenchymal tumors include five different histological types: pineocytoma (PC), pineal parenchymal tumors of intermediate differentiation (PPTID), papillary tumor of the pineal region (PTPR), pineoblastoma (PB), and desmoplastic myxoid tumor of the pineal region, SMARCB11-mutant; GCTs include germinoma, embryonal carcinoma, yolk sac tumor, choriocarcinoma, teratoma, and mixed GCTs.Neuroimaging assessment plays a critical role in the diagnosis, surgical planning, and follow-up of patients with pineal masses. Surgery can serve as the primary means of treatment, ranging from biopsy to total resection; however, pineal region tumors associated with obstructive hydrocephalus can be surgically treated via ventricular internal shunt or endoscopic third ventriculostomy. Radiotherapy remains a fundamental component of the multidisciplinary treatment approach for most pineal region tumors; however, treatment volumes depend on histological subtype, grading, extent of disease, and combination with chemotherapy.For localized germinoma, the current standard of care is chemotherapy followed by reduced-dose whole ventricular irradiation, along with a boost to the primary tumor. For pineoblastoma patients, postoperative radiation has been associated with higher overall survival. For other pineal tumors, the role of radiotherapy remains poorly studied and is usually reserved for aggressive (grade 3) or recurrent tumors. The use of systemic therapy mainly depends on histological and prognostic factors, such as residual disease and metastasis.For pineoblastoma patients, chemotherapy protocols are based on various alkylating or platinum-based agents, vincristine, etoposide, cyclophosphamide, and are used in association with radiotherapy. Regarding GCTs, their chemosensitivity is well known, and treatment is based on cisplatin or carboplatin, which may include etoposide, cyclophosphamide, or ifosfamide prior to irradiation. Similar regimens containing platinum derivatives are also used for non-germinomatous GCTs with very encouraging results. However, due to a deeper understanding of the biology of various molecular subtypes of the disease, new drugs based on targeted therapy are expected to emerge in the future.On behalf of the EURACAN domain 10 group, we review the most significant recent advances in the histopathological characteristics, neuroimaging assessment, and treatment of pineal region tumors.SummaryPineal region tumors are rare intracranial tumors. In recent years, there has been a deeper understanding of the molecular mechanisms of these tumors, leading to new classifications and potential systemic treatment methods. Surgery remains the primary means of treatment, while radiotherapy and systemic therapy depend on histology, molecular, and clinical features. This article focuses on the latest advances in the diagnosis and treatment of these tumors.1. IntroductionEURACAN is the European Reference Network for all rare adult solid cancers (http://euracan.eu, accessed on July 26, 2022). This network includes healthcare providers and patient representatives from across Europe, aiming to address rare adult solid cancers. Within the EURACAN network, there is a highly specialized group of physicians dedicated to treating “rare brain and spinal cord cancers” (referred to as Domain 10). On behalf of the EURACAN Domain 10 group, we provide an overview of regional pineal tumors.The pineal region is a composite anatomical space comprising the pineal gland and surrounding structures, including the epithalamus, quadrigeminal cistern, and posterior wall of the third ventricle. Pineal region tumors (PRTs) are rare, accounting for less than 1% of all intracranial tumor lesions, and are a group of histologically heterogeneous tumors, including primary pineal parenchymal tumors, germ cell tumors (GCTs), and tumors originating from adjacent structures, such as choroid plexus tumors, meningiomas, and gliomas. Lymphomas, atypical teratoid rhabdoid tumors (ATRT), and metastatic tumors, although uncommon, may also occur in this region. Pineal parenchymal tumors and germ cell tumors account for over 70% of all pineal region tumors. Treatment methods include biopsy and surgery, radiotherapy, and chemotherapy. However, due to the low incidence of these tumors, the different histological types, and the limited studies reported in the literature, treatment remains non-standardized.
| Pineal Region Tumors | |||
| Germ Cell Tumors | Pineal Parenchymal Tumors | Other Rare Tumors | Pineal Metastatic Tumors |
| – Germinoma– Embryonal Carcinoma– Yolk Sac Tumor– Choriocarcinoma– Teratoma (mature and immature)– Mixed Germ Cell Tumors | – Pineocytoma– Pineal Parenchymal Tumors of Intermediate Differentiation– Papillary Tumor of the Pineal Region (PTPR)– Pineoblastoma– Desmoplastic Myxoid Tumor of the Pineal Region (SMARCB11-mutant) | – Diffuse Gliomas– Focal Astrocytomas– Glioblastoma and Ependymomas– Embryonal Tumors– Meningiomas– Mesenchymal Non-Meningeal Tumors– Lymphomas |
Figure 1 Overview of Pineal Region Tumors According to WHO 2021 Classification.
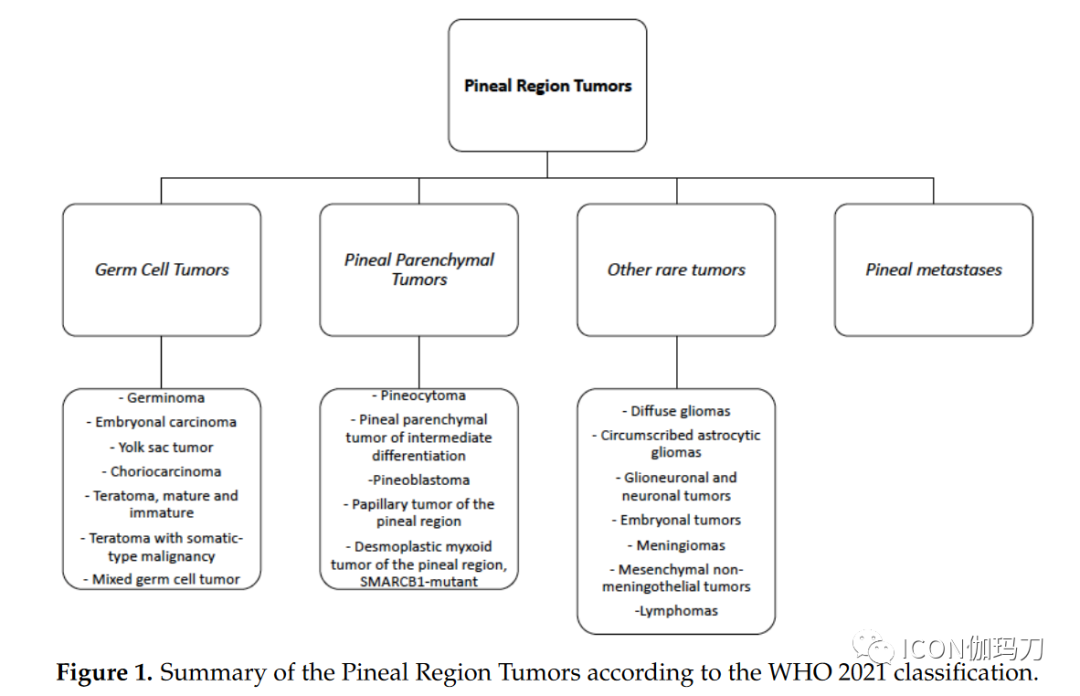
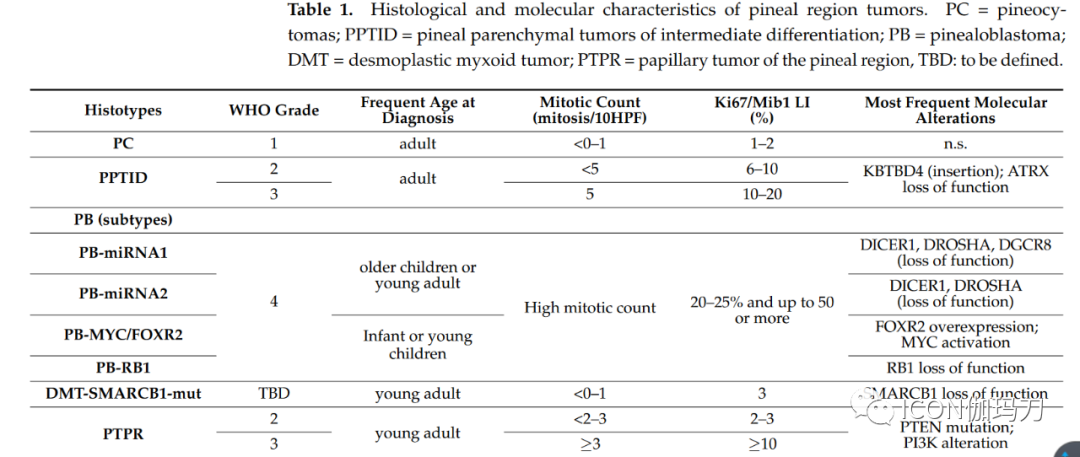
On behalf of the EURACAN domain 10 group, we review the most significant recent advances in the histopathological characteristics, neuroimaging assessment, and treatment of pineal region tumors.2. Pathological and Molecular FeaturesApproximately 30% of all pineal region tumors arise from pineal parenchyma. According to the World Health Organization (WHO) classification, pineal parenchymal tumors include four different histological types, stratified according to histological features and grade: pineocytoma (PC; WHO grade 1), pineal parenchymal tumors of intermediate differentiation (PPTID; WHO grades 2 and 3), papillary tumor of the pineal region (PTPR; WHO grades 2 and 3), and pineoblastoma (PB); most pineoblastomas (PB) occur in children, while pineocytomas (PC) and pineal parenchymal tumors of intermediate differentiation (PPTID) are more common in young adults. Additionally, the latest WHO 2021 classification of central nervous system (CNS) tumors confirms a new entity, the desmoplastic myxoid tumor (DMT), based on SMARCB11 mutations and lack of malignant histological markers.Table 1 Histological and Molecular Features of Pineal Region Tumors.PC = Pineocytoma; PPTID = Pineal Parenchymal Tumors of Intermediate Differentiation; PB = Pineoblastoma; DMT = Desmoplastic Myxoid Tumor; PTPR = Papillary Tumor of the Pineal Region; TBD: To Be Determined.
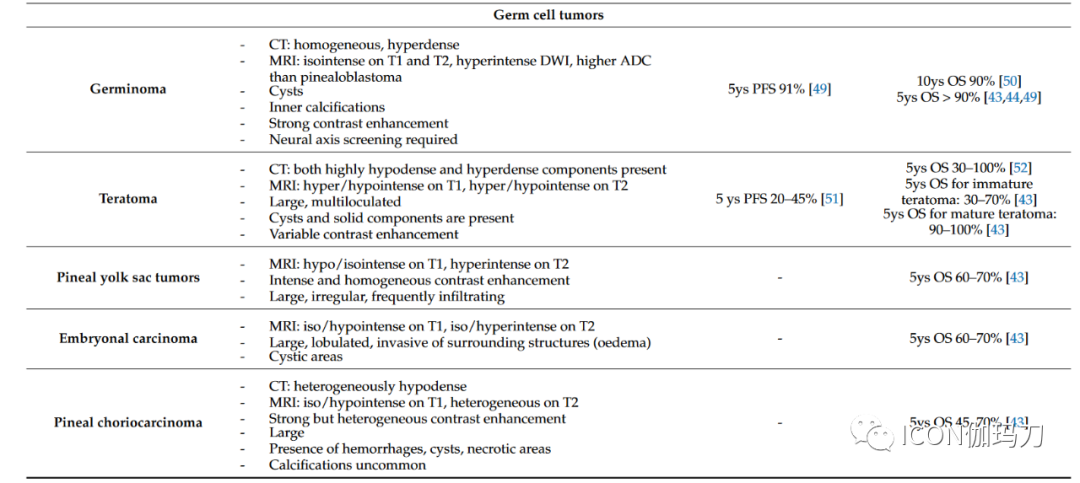
Pineocytoma (PC) accounts for about 25% of all primary pineal parenchymal tumors. PC is typically well-defined, slow-growing, and has a favorable prognosis, composed of well-differentiated cells resembling mature pineal cells, forming enlarged lobules. The tumor is non-invasive, rarely shows mitotic figures, and exhibits minimal nuclear atypia. Features of neuronal differentiation (ganglion cells) may be present. A distinct feature is the presence of pinocytomatous rosettes composed of neoplastic cells surrounding a central neuropil core. Tumor cells exhibit diffuse immunoreactivity for synaptophysin and neurofilaments. The proliferation rate measured by the percentage of tumor cells labeled by Ki-67/Mib1 is low (averaging 1 – 2%). There is limited molecular research on PCs, primarily focused on cytogenetic alterations involving chromosomes X, 5, 8, 11, 14, 19, and 22.Due to the lack of strict histopathological classification criteria and the presence of histological features ranging from well-differentiated PCs (pineocytoma) to high-grade PBs (pineoblastoma), the diagnosis of PPTID is challenging. PPTID accounts for approximately 40 – 45% of all primary pineal parenchymal tumors. PPTID exhibits heterogeneous structures, lacking the primitive small round cell appearance seen in PBs, and endothelial cell proliferation or necrosis is rarely detected based on grading. WHO grade 2 PPTID typically maintains high neurofilament expression, similar to PC, with low mitotic counts (<5 mitoses per 10 HPF), but the Ki67/MIB-1 labeling index is generally higher than that of PC, ranging from 6% to 10%. WHO grade 3 PPTID exhibits a diffuse growth pattern, lacking PC-like areas, with absent or very limited neurofilament expression, and gradually acquires a dedifferentiated phenotype. Therefore, mitotic counts are higher (>5 mitoses per 10 HPF), with Ki67/MIB-1 labeling indices ranging from 10-20%. Endothelial cell proliferation and necrotic foci are also common. Unlike PC, PPTIDs, especially WHO grade 3 PPTIDs, show an increased rate of molecular alterations. Recently, overexpressed genes such as PRAME, CD24, POU4F2, and HOXD13 have been reported in grade 3 PPTID, which may serve as potential useful biomarkers in differential diagnosis with grade 2 PPTID. Additionally, high expression of CD24 and PRAME may have prognostic value as it is associated with significantly shortened survival. In PPTID, gains of chromosomes 4q and 12q and losses of chromosome 22 have been reported as frequent chromosomal alterations. Recent advancements in genomic and transcriptomic analyses have made it possible to describe the carcinogenic driving factors. KBTBD4 intron insertions are common molecular alterations in PPTID. In contrast, DICER1 and DROSHA mutations are limited to PBs and may assist in differential diagnosis. ATRX mutations have also been reported with protein loss.Pineoblastomas (PBs) account for approximately 30% of all pineal parenchymal tumors and about 10% of all pineal region tumors; they primarily occur in children or infants. PBs are composed of poorly differentiated and immature cells, exhibiting rapid growth and a tendency for leptomeningeal dissemination. Histologically, PBs show marked hypercellularity composed of small round immature cells with hyperchromatic nuclei frequently showing anaplasia, with frequent mitotic figures, apoptotic bodies, nuclear molding, necrosis, and a high Ki67/Mib1 labeling index ranging from 20–25% and up to 50% or more. Homer–Wright rosettes may also appear in the tumor. Most retain synaptophysin expression, while neurofilaments are typically absent, and GFAP may be partially positive. Diagnosis is challenging, especially in small biopsies, as it may be difficult to distinguish PBs from other malignancies occurring in the region, including GCTs (germ cell tumors), ATRT (atypical teratoid rhabdoid tumors), high-grade gliomas, and WHO grade 3 PPTIDs (pineal parenchymal tumors of intermediate differentiation). PBs frequently exhibit high expression of molecules such as UBEC2, SOX4, TERT, TEP1, PRAME, CD24, POU4F2, and HOXD13. CRX is a major transcriptional regulatory factor expressed in the pineal gland and retina, and its expression level helps support the pineal lineage. DNA methylation profiles reveal four clinically relevant and biologically distinct PB subgroups, which differ in diagnostic age, metastatic tendency, molecular alterations, and clinical outcomes. Recurrent homozygous deletions and mutations of microRNA processing pathway genes (DICER1, DROSHA, and DGCR8) define the PB subtypes of microRNA processing alteration group 1 and microRNA processing alteration group 2, both of which frequently occur in older children (ages 3 – 18) with intermediate to good prognosis (5-year OS between 70 – 100%). Notably, the PB microRNA processing alteration group is the most common CNS malignancy in patients carrying germline DICER1 mutations. Overexpression of FOXR2 and activation of MYC characterize a more aggressive PB subgroup with MYC/FOXR2 alterations, primarily occurring in infants. Loss of RB1 gene function is characteristic of the PB RB1 alteration subgroup, primarily originating in poor prognosis, rapidly progressing infants (median age 1-2 years). Cytogenetic alterations are also encountered, with frequent gains of chromosomes 1, 6, 7, 12, and 17, and losses of chromosomes 8, 14, and 16. Interestingly, loss of chromosome 16q is significantly associated with poorer prognosis in PBs with MYC/FOXR2 alterations and RB1 alterations. Therefore, methylation analyses reveal considerable molecular heterogeneity associated with different clinical features and survival rates in different PBs. In this regard, molecular grouping is crucial for guiding future clinical research and patient stratification for personalized treatment.The DMT SMARCB11 mutation (desmoplastic myxoid tumor SMARCB1 mutation) in the pineal region is a newly discovered rare entity, primarily affecting young individuals and exhibiting distinct clinical and histopathological features. Histologically, DMT SMARCB11 mutation is composed of small to medium-sized cells and irregularly shaped elongated blood vessels within a loose basophilic myxoid matrix. Focal calcifications may be present, with low mitotic activity and no tumor necrosis. All patients show nuclear SMARCB11/INI1 protein expression loss and have an immune response to EMA and CD34. In most patients, the Ki67/MIB1 proliferation index is low (median 3%). Interestingly, the DMT SMARCB11 mutation in the pineal region shares a close methylation profile with ATRT-MYC, a recently identified ATRT methylation molecular subgroup carrying SMARCB11 deletions, with relatively good prognosis.Pineal region papillary tumors (PTPR) are rare WHO grade 2 or 3 pineal tumors characterized by morphological changes with an epithelial appearance, a predominantly papillary architecture reminiscent of ependymomas, and the presence of ependymal rosettes. Despite its anatomical association with the pineal gland, a PTPR is believed to originate from specialized cells in the posterior third ventricle near the pineal gland that show ultrastructural features of ependymal and neuroendocrine differentiation. PTPR typically expresses cytokeratins (including CK18) as well as S100, Vimentin (wave protein), and neuron-specific enolase (NSE), but does not express neurofilaments, while epithelial membrane antigen (EMA) and GFAP (glial fibrillary acidic protein) expression is inconsistent. Mitoses are uncommon, but necrosis is frequently observed. Increased mitoses (≥3 mitoses per 10 high power fields [HPF]) and proliferative activity (Ki67/MIB1 index ≥10%) as well as loose papillary structures have been shown to be associated with shorter progression-free survival. Almost all PTPR patients exhibit chromosomal deletions associated with PTEN gene mutations and PI3K pathway activation. Other changes include frequent losses of chromosomes 3 and 22q, as well as gains of chromosomes 8p, 8, and 12. FOXJ1 nuclear expression is often expressed in normal ependymal cells and ependymomas, which can help differentiate from histopathological mimics, particularly PPTID (pineal parenchymal tumors of intermediate differentiation).Finally, germ cell tumors (GCTs) in the pineal region account for 50 – 60% of pineal tumors. GCTs are more common in young individuals and children, primarily affecting male patients. Germinoma is the most common type of pineal tumor, but all different histological types may exist, including choriocarcinoma, teratoma, embryonal carcinoma, yolk sac tumor, and mixed germ cell tumors. Other tumors, including metastatic cancers that spread to the pineal gland, are extremely rare.3. Neuroimaging AssessmentNeuroimaging plays a critical role in the diagnosis, surgical planning, and follow-up of patients with pineal masses. Computed tomography (CT) and magnetic resonance imaging (MRI) can provide useful information regarding the location, size, and shape of pineal tumors. While MRI remains the preferred tool for tumor characterization, brain CT is often the first-line imaging method for patients with rapidly deteriorating neurological conditions or gradually worsening headaches. CT is sufficient for detecting pineal region tumors or hydrocephalus secondary to compression of the tectum of the midbrain and obstruction of the aqueduct. Additionally, CT is the preferred method for assessing calcifications and hemorrhages. MRI best displays lesion enhancement patterns, the presence of associated lesions in the suprasellar region, or the presence of leptomeningeal dissemination. Furthermore, MRI can better differentiate tumors from other benign pineal region masses, such as pineal cysts, epidermoid cysts, dermoid cysts, or arachnoid cysts.Overall, the neuroimaging literature clearly describes most types of pineal region tumors, outlining their main morphological and signal or density characteristics (see below). However, the cellular diversity of the region and the variability of brain structures, along with the general overlap in neuroimaging features among different tumor types, make differential diagnosis somewhat challenging. In most cases, the final diagnosis may depend on clinical data (age and sex) and imaging findings, but ultimately relies on laboratory serum or cerebrospinal fluid tumor markers, as well as histological and immunohistochemical examinations.Although neuroimaging often cannot determine the nature of the lesions, it is essential for managing pineal mass lesions, distinguishing benign lesions from tumors, narrowing the differential diagnosis for pineal region tumors, determining the relationship with vascular structures (such as intracranial veins) and parenchymal structures, detecting leptomeningeal dissemination or regional dissemination, or the presence of other concomitant lesions. Additionally, neuroimaging has become an indispensable tool for postoperative lesion follow-up, usually arranged based on the clear characteristics of histological lesions. CT is often used in the early postoperative period to rule out surgery-related complications and determine the severity of hydrocephalus. However, subsequent neuroimaging, typically using contrast-enhanced MRI, is performed to assess the entire neuroaxis that is included.The following are radiological descriptions of the three main types of pineal region tumor masses: parenchymal tumors, germ cell tumors, and supportive tissue tumors (gliomas). Metastatic, vascular, and benign lesions have also been reported.3.1. Pineal Parenchymal Tumors– Pineocytoma typically appears as a well-defined tumor, with a maximum size of less than 3 cm. Compared to the adjacent brain, the lesion appears isodense/hyperdense on CT, low/equal signal on T1, and isointense on T2. The tumor tends to be solid and exhibits strong homogeneous enhancement. Cystic changes may be present, which in some cases may make it difficult to distinguish from pineal cysts; nodular or thickened wall enhancement aids in determining its tumor nature. Hemorrhage within the lesion may occur, while calcifications are usually peripheral, distinguishing pineocytoma from pineal germ cell tumors, which tend to engulf pineal calcifications. Pineal stroke has been reported.– Pineoblastoma typically presents as a larger (greater than 3 cm), irregular, poorly defined lobulated tumor, often invading adjacent brain. On CT, due to high cell density, it appears hyperdense compared to the adjacent brain, frequently exhibiting necrotic areas and hemorrhagic changes due to its high malignancy. Like pineocytoma, calcifications are dispersed peripherally (“blasted calcifications”). Cystic appearances may occur, but the walls are usually thicker and more irregular than those of pineal cysts. On MRI, pineoblastoma exhibits heterogeneous signal intensity (low to equal signal on T1, equal to high signal on T2), with restricted diffusion. In addition to elevated choline and decreased N-acetylaspartate, MR spectroscopy shows slightly elevated peaks of glutamate and taurine (approximately 3.4 ppm). Contrast enhancement is clear, and 15% of patients present with CSF dissemination at diagnosis, rising to 45% during the disease course. Therefore, screening of the entire neuroaxis is necessary during biopsy diagnosis/imaging suspicion and follow-up. Obstructive hydrocephalus is common at onset.– Pineal parenchymal tumors of intermediate differentiation range from well-defined tumors with pineocytoma-like features to poorly defined, rapidly growing and/or aggressive masses with low ADC values. T2 often demonstrates isointense signals, may exhibit cystic areas and heterogeneous enhancement. Since these tumors may disseminate along the cerebrospinal fluid, enhanced MRI examinations of the entire craniospinal axis are necessary.– Papillary tumor of the pineal region appears as a well-defined T2 hyperintense mass on MRI, accompanied by varying degrees of contrast enhancement. Cystic lesions are common, and their protein content may lead to uneven T1 signals. Due to the potential for local tumor recurrence and leptomeningeal tumor dissemination, MRI enhancement scans of the entire craniospinal axis are recommended.3.2. Germ Cell TumorsGerm cell-derived tumors include germinoma, teratomas, and less commonly embryonal carcinoma, pineoblastoma yolk sac tumors, and choriocarcinoma.– On CT, pineal germinoma typically presents as a uniformly hyperdense mass compared to the adjacent brain. Cysts are present in 20-52% of cases. Internal calcifications are common, usually representing normal pineal calcifications being engulfed by the tumor. On MRI, germinoma typically shows isosignal on T1 and T2 similar to gray matter, with relatively high DWI signal, and ADC values are usually higher than those of pineoblastoma. Contrast enhancement is vivid (Contrast enhancement is vivid). At onset, disseminated disease along the third ventricle is common, with subependymal tumors (13%).– Teratomas typically present as large, multiloculated, lobulated lesions, including cystic and solid components, as well as areas of fat, calcification, and liquid lesions within the mass. On CT, most teratomas show at least some highly low-density or high-density components (respectively fat and calcification). On MRI, teratomas may exhibit T1 hyperintense components due to fat and protein/lipid-rich fluids, and T1 low signal components due to calcifications and blood products. Due to the wide variability in tissue components, T2-weighted imaging also tends to be heterogeneous, with low or high signal components within the tumor. After the injection of contrast agents, solid components exhibit varying degrees of enhancement. Immature teratomas are more homogeneous, with irregular infiltrative margins, and fewer cysts and calcifications, making them more challenging to differentiate from other pineal tumors. Secondary malignant tumors are not uncommon; therefore, monitoring for secondary malignant tumors and growing teratoma syndrome is recommended.– Pineal yolk sac tumors are rare and seem indistinguishable from other germ cell tumors. These lesions are usually larger, irregular, and often infiltrate adjacent structures; they present with varying densities and calcifications. On MRI, lesions show low signal on T1 and high signal on T2. Contrast enhancement is strong and homogeneous.– Embryonal carcinoma usually presents as large masses, showing low signal on T1 and high signal on T2 compared to gray matter. Their margins are lobulated and irregular; tumor margins with edema suggest invasion of adjacent structures. Cystic areas are common; solid portions show vivid contrast enhancement.– Pineal choriocarcinoma is a large, highly vascular lesion with a tendency for bleeding (as do its metastases). The mass typically appears oval with well-defined margins, but irregular infiltrative margins can be observed. Most lesions show isosignal on T1 and heterogeneous on T2 compared to the cortex. Contrast enhancement is usually prominent but uneven. Microcysts and large cysts or necrotic areas and mild to moderate edema around the tumor are common. Hemorrhage leads to uneven signals, blooming on T2* and SWI, with high T1 signal focal areas. CT shows the tumor as heterogeneous low-density; calcifications are rarely seen. The most common radiological features and survival outcomes of the most common pineal parenchymal tumors and germ cell tumors are summarized in Table 2.Table 2 Summary of Radiological Features and Survival Outcomes of the Most Common Pineal Parenchymal Tumors and Germ Cell Tumors.

3.3. Other Tumors of the Pineal RegionThe diversity of brain structures in the pineal region leads to a multitude of possible tumor types (such as meningiomas, gliomas, melanomas, ependymomas, etc.). Additionally, metastatic tumors may also occur in the pineal region.– Pineal meningiomas are well-defined masses, mostly arising from the contiguous tentorium, the tela choroidea, or the velum interpositum. On CT, compared to gray matter, lesions usually show isosignal or high signal; calcifications can be detected in 15-20% of patients. MRI signals are variable. The mass shows strong homogeneous contrast enhancement, typically involving adjacent dural structures (dural tail sign).– Primary pineal melanomas are extremely rare and may present with pigmented or unpigmented MRI patterns. The former is more common, with the mass appearing high signal on T1 due to the presence of melanin, low signal on T2, or isosignal. Unpigmented MRI presentations are less common, with lesions showing low or isosignal on T1 and high signal on T2 (less than 10% of cells contain melanin). Typical presentations include strong contrast enhancement.– Ependymomas present as heterogeneous lesions, typically low signal to isosignal on T1 and isosignal to high signal on T2. Cysts, calcifications, and hemorrhages are common. Contrast enhancement may be present; diffusion restriction is uneven, but due to relatively low cell density, ADC is predominantly high.– Pineal gliomas include pilocytic astrocytomas, IDH-mutant astrocytomas, oligodendrogliomas, glioblastomas, and choroid plexus papillomas, although well-differentiated astrocytomas are the most common. In addition to tumors from the pineal gland, gliomas in the pineal region also include parietal gliomas, thalamic gliomas, and splenial gliomas. Therefore, the imaging features vary considerably based on tumor grading and anatomical origin. There are different manifestations of necrosis, calcification, cysts, and hemorrhage within lesions; contrast enhancement is also variable.– Patients with any history of malignant tumors should consider pineal metastatic tumors. Although lung cancer is the most common, pineal metastases from almost all malignancies have been reported. On neuroimaging, metastatic lesions often appear isodense to gray matter. Lesion margins may be well-defined or infiltrate adjacent tissues. Strong contrast enhancement is typical; necrosis and cystic changes may occur. Leptomeningeal dissemination is observed in two-thirds of patients; therefore, enhanced MRI examinations of the entire craniospinal axis are required.3.4. Non-Tumorous and Benign Lesions of the Pineal RegionSeveral conditions can be observed in this complex region.– Pineal cysts are very common in the general population (up to 40% in autopsy series) and are almost always asymptomatic. On MRI, benign pineal cysts typically appear oval, thin-walled, unilocular or multilocular, or filled with protein or hemorrhagic fluid, usually showing no restriction on DWI. MRI shows that compared to gray matter, cysts exhibit low signal to isosignal on T1, and isosignal to high signal on T2. A common feature is a thin rim of calcification (in 25% of patients), and most importantly, wall enhancement is smooth (i.e., non-nodular) and usually < 2mm thick. Due to gadolinium infiltration into cystic fluid, delayed imaging may reveal internal enhancement. There is no invasion of adjacent structures and minimal or no mass effect. The cerebral aqueduct remains patent.– Velum interpositum cysts are a common anatomical variant, accompanied by normal expansion of the velum interpositum (cistern of the velum interpositum) (diameter >1 cm). Cysts typically appear triangular, pointing forward, and typically communicate with the cerebral veins along their lateral walls. CT and MRI show cyst contents characteristic of cerebrospinal fluid.– Arachnoid cysts present as significant extraxial fluid collections, with signals similar to cerebrospinal fluid on all MRI sequences, including DWI. No enhancement is seen after contrast.– Epidermoid cysts are non-tumorous lesions, typically appearing as low-density on CT; focal calcifications may be observed. On MRI, these masses usually exhibit CSF-like signals on T1 and T2, but show bright signals on DWI. Typical contrast enhancement is absent, although faint, very delayed peripheral enhancement may be observed.– Dermoid cysts are usually well-defined round midline masses, typically appearing as significantly low-density on CT, high signal on T1, while showing low signal on fat-suppressed T1 due to the fat content within the cyst. Calcifications may be present within the wall. Typically, dermoid cysts do not enhance after contrast injection, although a thin peripheral rim may be observed. Rupture of the cyst may release fat droplets into the subarachnoid space, aiding in distinguishing lipomas from mature teratomas.– Pineal lipomas are easily identified on CT due to their very low fat density. Calcifications may be present. On MRI, well-defined lesions exhibit uniform high signal on T1 and low signal on fat suppression imaging. The typical absence of enhancement helps distinguish lipomas from mature teratomas.– Vein of Galen aneurysm malformation is a congenital arteriovenous malformation leading to abnormal dilation of the vein of Galen, usually observed in neonatal or fetal imaging. This condition is easily identified by magnetic resonance angiography and due to the flow void signal of blood flow.– Cavernous malformations are rare, but their MRI features typically allow for rapid diagnosis in most patients. On T2 images, they often present as “pop-corn like” appearances, surrounded by low signal hemosiderin rings. MRI signal intensity depends on whether there has been recent hemorrhage or thrombosis.4. Role of SurgeryAdvanced microsurgical techniques (endoscopy, neuronavigation, electrophysiological monitoring) combined with significant improvements in anesthesia and recovery management have facilitated the surgical management of pineal region tumors (PRTs). With the exception of germ cell tumors and lymphomas, maximal microsurgical resection remains the gold standard for PRTs.4.1. Management of HydrocephalusMore than half of PRT patients present with obstructive hydrocephalus at diagnosis. In such cases, timely discussion of hydrocephalus management is warranted, and ventricular internal shunt or endoscopic third ventriculostomy (ETV) may be employed. The latter is preferred because, in addition to relieving hydrocephalus, when the tumor bulges in the posterior part of the third ventricle, it provides an opportunity for biopsy with a sensitivity greater than 90%, primarily due to the significantly lower incidence of complications associated with hemorrhage from the highly vascular tumor being less than 3%. Typical ventricular shunts (ventricular-peritoneal shunt or ventricular-atrial shunt) also carry the risk of tumor dissemination. Systematic collection of cerebrospinal fluid samples for PRT marker analysis and tumor cell screening is performed. During follow-up, approximately 15% of patients treated with ETV may require internal ventricular shunting due to third ventricle stoma dysfunction (approximately 15% of patients who have been treated with ETV may require an internal ventricular shunt because of third ventricle stoma dysfunction).4.2. BiopsyBefore multidisciplinary treatment discussions, obtaining tissue samples is crucial. In most patients with hydrocephalus, biopsy may be possible during endoscopic third ventriculostomy, especially for large tumors extending anteriorly into the third ventricle (Figure 2A). In other patients, stereotactic biopsy is usually performed under neuronavigation, providing histological diagnosis in 87.97% of patients (Figure 2B-D). The disadvantage of biopsy in PRTs remains the risk of obtaining non-representative samples in mixed tumors containing different tumor types. Despite the proximity to the venous complex anatomy (the vein of Galen and its tributaries), the morbidity and mortality rates associated with PRT biopsies remain similar to other brain regions (mortality rate 0.2%, transient morbidity rate 8.4%, and final morbidity rate less than 1.2%).
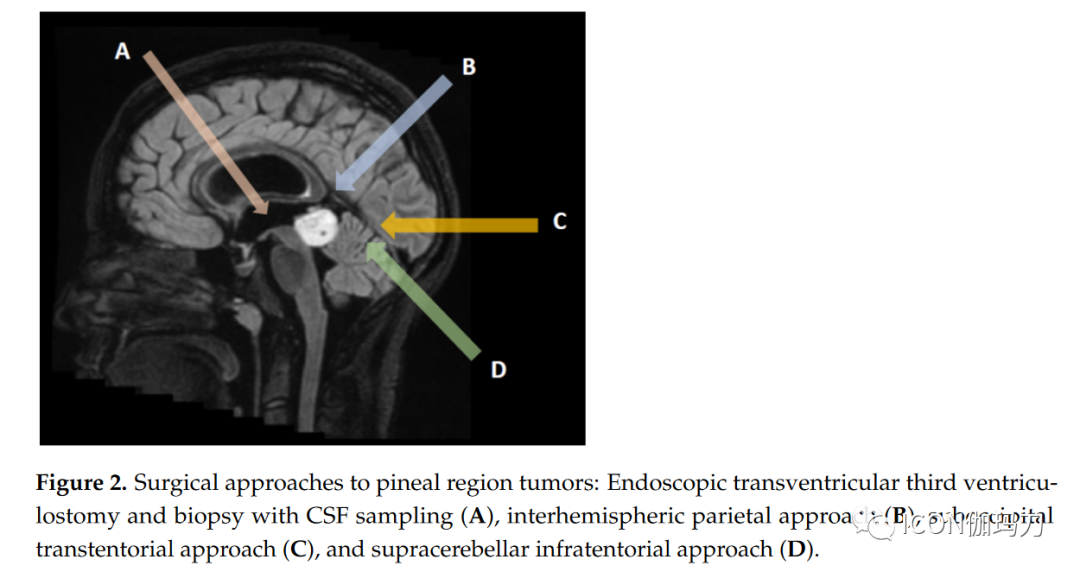
Figure 2 Surgical approaches to pineal region tumors: endoscopic third ventriculostomy and cerebrospinal fluid sampling biopsy (A), hemispheric parietal interhemispheric approach (B), suboccipital approach (C), and suboccipital supratentorial approach (D).4.3. Surgical ResectionWhen blood and/or cerebrospinal fluid markers are negative, surgical resection of PRT remains the standard method and should be discussed in multidisciplinary meetings involving neuro-oncologists, radiation oncologists, and neurosurgeons. Due to the deep and critical venous relationships of PRTs, surgical management should be conducted at tertiary centers with extensive experience in these tumors. In a large cohort of patients, the surgical mortality rate is less than 3%, but the complication rate can be as high as 20%, particularly in patients with ocular movement abnormalities and visual field defects. The specific choice of surgical approach depends on the relationship of the tumor to the vein of Galen complex and the surgeon’s experience. For tumors extending upward and displacing the venous complex downward, the suboccipital approach is preferred (Figure 2C). In the absence of an open foramen ovale, patients are typically placed in a sitting position; otherwise, a three-quarters prone position (Park Bench) is used. For right-handed patients, the craniotomy should expose the right lateral sagittal sinus and superior sagittal sinus. The occipital lobe is then carefully dissected and retracted, exposing the falx and dura mater angle and the straight sinus. The tentorium dura mater is then incised parallel to the straight sinus 1 cm until reaching the thick layer of arachnoid in the pineal region. Care should be taken to preserve all venous tributaries of the vein of Galen while resecting and debulking the tumor. The tumor is separated from the superior cerebellum and the tectum until entering the third ventricle cavity. The lateral oculomotor nerve may be encountered and preserved. Due to occipital lobe retraction, this approach carries a significant risk of visual field dysfunction, which is usually transient but, unfortunately, can sometimes be permanent. The suboccipital supratentorial approach (Figure 2D) provides a direct route to the pineal region, particularly for tumors occurring beneath the venous complex. To achieve optimal exposure, patients should be positioned as upright as possible to allow the cerebellum to naturally descend with gravity. Sometimes, it may be necessary to sacrifice one or two bridging veins connecting the superior surface of the cerebellum and the tentorium. The approach may be slightly lateral to the midline to avoid damaging the midline bridging veins. This approach provides a direct path to the pineal region beneath the vein of Galen complex. The main potential complications are small cerebellar damage caused by venous infarction or bruising during retraction, as well as occipital dysfunction (oculomotor dysfunction) and pneumatocele. Depending on the tumor’s extension laterally or anteriorly into the third ventricle, other approaches may be employed. Notable lateral extensions can be managed through a trans-cortical approach via the parietal bone, but the risk of visual field complications is high. Anterior extensions into the third ventricle can be accessed through a transventricular approach (through the foramen, dome…), with a significant risk of memory deficits. In a recent retrospective study, Shepard et al. analyzed surgical and tumor outcomes in 68 patients who underwent surgery for pineal region tumors. The mean age at surgery was 30.9 ± 15.3 years, with 83.8% of patients presenting with preoperative hydrocephalus and a median preoperative Karnofsky performance status (KPS) of 90 points. Germ cell tumors accounted for 20.6%, and pineal parenchymal tumors accounted for 30.9%. Gliomas comprised 32.4% of the lesions. In this study, 52.9% of patients achieved gross total resection (GTR), with higher resection rates for low-grade tumors. In terms of adverse events, 45.6% of patients experienced one or more adverse events within 30 days postoperatively; the most common adverse events were new or worsening hydrocephalus (14.7%) and postoperative focal motor deficits (10.3%). The 30-day mortality rate was 5.9%, primarily due to intratumoral hemorrhage (3 out of 4 cases). GTR, low-grade tumors, and improvement in general performance status were associated with improved overall survival. Due to the small number of patients, the impact of histopathology on overall survival was not analyzed.4.4. Specific Treatment of Pineal CystsPineal cysts are common benign expansile cysts of the pineal gland, found in up to 5% of brain imaging. Most are less than 10mm and asymptomatic, but they can rarely enlarge and cause aqueductal stenosis and secondary hydrocephalus. They may also present with headaches, ocular movement disturbances due to tectal compression, or cerebellar symptoms. Benign pineal cysts typically appear as T1 low signal, T2 high signal cystic lesions with thin wall micro-enhancement. Thicker walls and/or associated solid components should be considered tumors and managed as cystic pineal region tumors. Asymptomatic benign pineal cysts should be followed annually to ensure no progression. Management of symptomatic patients remains controversial. In patients with benign pineal cysts leading to symptomatic hydrocephalus, a commonly used option is endoscopic third ventriculostomy, which can also allow for biopsy of the cyst wall and fenestration into the third ventricle cavity. For symptomatic patients without hydrocephalus (oculomotor signs or cerebellar signs), direct microsurgical approaches may also be recommended.4.5. Role of RadiotherapyRadiotherapy remains an important component of the multidisciplinary treatment approach for intracranial germ cell tumors. In recent decades, significant advancements in radiotherapy techniques have made treatment more precise and effective. Currently, advanced external beam techniques include image-guided radiotherapy (IGRT), intensity-modulated radiotherapy (IMRT), stereotactic radiosurgery (SRS), and volumetric modulated arc therapy (VMAT). Compared to three-dimensional conformal radiotherapy techniques, modern techniques provide highly conformal dose distributions, improving target coverage and sparing normal tissues, potentially reducing the risk of long-term sequelae, particularly neurocognitive dysfunction. Additionally, there is increasing interest in proton therapy; for larger intracranial lesions, it offers a more favorable dose distribution for surrounding normal tissues than photon therapy. Treatment volumes include craniospinal irradiation, whole brain irradiation, whole ventricular irradiation, or focal radiotherapy. These volumes depend on histological subtype, grading, extent of disease, and combination with chemotherapy. Current radiotherapy indications for different tumor types are shown in Table 3.For germ cell tumor patients, craniospinal irradiation of 36 Gy followed by a boost of 50-54 Gy to the primary tumor, delivered in 1.8 Gy fractions, has long been considered the standard treatment method prior to the 1990s, with reports of over 90% 5-year event-free survival. Given the generally favorable prognosis for localized germ cell tumors and concerns about the long-term toxicity of radiotherapy (including secondary malignancies, strokes, and declines in neurocognitive ability), clinical treatment has shifted toward chemoradiotherapy, which includes lower radiation doses and smaller target volumes. For localized germ cell tumors, the current standard of care is chemotherapy followed by reduced-dose whole ventricular irradiation of 24 Gy, plus a boost of 16 Gy to the primary tumor (1.8 Gy per fraction), with lower doses of 18 Gy and 12 Gy for patients achieving complete remission after chemotherapy, as recommended by the pediatric oncology group.Table 3 Radiotherapy for Pineal Region Tumors.
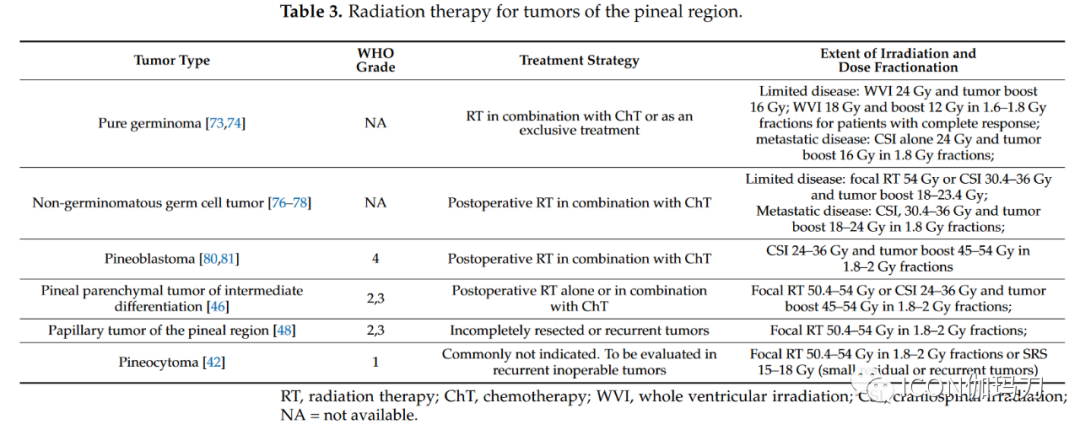
Similarly, bifocal germ cell tumors can also be treated with whole ventricular irradiation and primary boost therapy. Whole ventricular irradiation is currently recommended, as using local field radiotherapy may lead to excessive recurrence in the ventricles. When radiotherapy is used as a single modality, the treatment scope includes whole ventricular radiotherapy at a dose of 24 Gy and tumor boost therapy at a dose of 40-45 Gy (1.8 Gy per fraction). For patients with disseminated disease, treatment requires craniospinal irradiation of 30.4-36 Gy, delivered in 18-20 daily fractions, with a boost of 40-45 Gy to the primary and metastatic sites at a dose of 1.8 Gy per fraction; however, in patients receiving combined chemoradiotherapy, craniospinal doses and tumor boost doses can be reduced. The majority of evidence regarding the effects of radiotherapy on pineal tumors comes from retrospective studies and small case series, making it challenging to define standard treatment. Pineoblastoma is more common in pediatric patients than in adult patients. In the pediatric population, postoperative radiotherapy is associated with higher overall survival; in contrast, there is a lack of high-quality data on the effects of radiotherapy on adult pineoblastoma. Among 213 adult pineoblastoma patients monitored in the Surveillance, Epidemiology, and End Results (SEER) database from 1975 to 2016, postoperative radiotherapy (hazard ratio 0.43, p < 0.05) and postoperative combined chemoradiotherapy (hazard ratio 0.38, p < 0.05) were independent prognostic factors affecting survival. In a comprehensive review of 109 studies, Tate et al. found that the overall survival rate was 54% during an average follow-up of 31±1.9 months (range 1-159 months). Adjuvant radiotherapy was not associated with improved survival after gross total resection, although post-radiotherapy subtotal resection improved survival; the 2-year survival rate after subtotal resection was 53%, and the 2-year survival rate after adjuvant radiotherapy was 64% (p < 0.05). Although the efficacy of radiotherapy remains controversial, it is currently recommended to combine platinum-based systemic chemotherapy with craniospinal irradiation (24-38 Gy) along the entire axis and (45-54 Gy) irradiating the tumor, delivered in fractions of 1.8-2.0 Gy. For other pineal tumors, including pineal parenchymal tumors of intermediate differentiation, papillary tumors of the pineal region, and desmoplastic myxoid tumors with SMARCB1 mutations, the clinical behavior and histological grading criteria remain unclear, and the role of postoperative adjuvant therapy remains uncertain. Currently, the indications for radiotherapy are primarily based on small retrospective studies and are generally only applicable to aggressive (grade 3) or recurrent tumors, while less aggressive tumors, such as grade 1-2 tumors, require observation. Pineal parenchymal tumors belong to intermediate differentiation, classified as grade 2 or 3 according to the WHO classification of CNS tumors, are rare tumors originating from pineal parenchyma, predominantly found in adults with limited aggressive behavior, positioned between pineocytomas and pineoblastomas (WHO 2021). Although many centers recommend adjuvant radiotherapy, the best treatment approach for these tumors remains unclear. In a patient-level analysis involving 127 patients across 29 studies, Mallick et al. reported that patients who received craniospinal irradiation or local radiotherapy had a higher survival rate compared to those who did not receive radiotherapy (252 months vs 168 months; P = 0.009), with no differences between different radiation methods. Although the management of these tumors remains unclear due to a lack of evidence, adjuvant radiotherapy is generally recommended for all patients with partial or subtotal resection of pineal parenchymal tumors. The local radiation dose using modern conformal techniques should be 50.4-54 Gy to limit the toxicity associated with long-term treatment. Grade 3 tumors typically recommend craniospinal irradiation. As for other pineal tumors, the treatment of papillary tumors of the pineal region remains unclear, with no standard guidelines. In a recent review of 77 articles involving 177 patients with papillary tumors of the pineal region, Yamaki et al. observed a 3-year survival rate of 83.5%, with a recurrence rate of 56.8% after a median of 29 months. Surgical resection was associated with survival, although the extent of resection did not affect prognosis. Adjuvant therapies, including radiotherapy (44%), chemotherapy (10.3%), and radiosurgery (10.8%), did not improve survival. Multivariate analysis revealed that tumor size and surgical treatment were associated with survival. Whenever feasible, complete surgical treatment of pineocytoma is recommended, with no further adjuvant therapy required. Clark et al. found in a literature review involving 64 articles and 166 patients that simple subtotal resection and subtotal resection plus radiotherapy showed no significant differences in progression-free survival. The 5-year survival rate is approximately 86%, with local recurrence being reported, and even rare cases of cerebrospinal fluid metastases. Some studies have evaluated second-course radiotherapy for recurrent tumors in the pineal region, although the efficacy and risk of toxicity remain controversial.6. Role of Systemic TherapyChemotherapy is primarily used as first-line treatment for pineal tumors, depending on histology and prognostic factors such as residual disease and metastasis. In this section, we will specifically review the chemotherapy methods for pineoblastoma and germ cell tumors (see Table 4). For other indications, chemotherapy regimens are similar to other localized recommendations.Table 4 Summary of Clinical Trials.
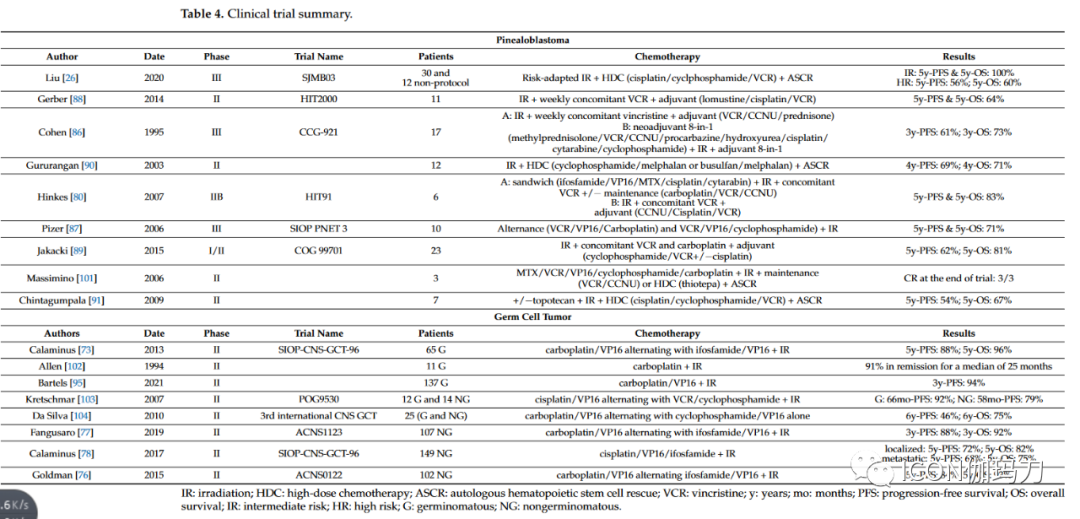
6.1. PineoblastomaThe chemotherapy regimens for adult pineoblastomas primarily stem from pediatric clinical trials. For non-metastatic patients, various chemotherapy regimens combined with radiotherapy yield a 5-year PFS rate of 60-70%. The use of chemotherapy alone is not recommended, and the prognosis is extremely poor. Chemotherapy protocols are based on various combinations of alkylating agents or platinum agents, vincristine (VCR), etoposide, and cytarabine. In the CCG-921 trial, randomized chemotherapy was used after craniospinal irradiation, whether with high-dose steroids, lomustine (CCNU) and VCR, or “eight-drugs-in-one-day” (methylprednisolone, VCR, CCNU, procarbazine, hydroxyurea, cisplatin, cytarabine, and cyclophosphamide), with a 3-year OS rate of 73%. In the SIOP PNET 3 study, alternating treatment with VCR, VP16, and carboplatin or cyclophosphamide combined with irradiation resulted in a 5-year OS rate of 71%. In the HIT91 trial, among children aged over 3 years receiving sandwich chemotherapy or adjuvant radiotherapy at the end of the study, 5 out of 6 survived without disease, with a median survival of 8.8 years. The HIT2000 trial evaluated the outcomes of patients receiving radiotherapy after adjuvant chemotherapy based on lomustine, cisplatin, and VCR, with a 5-year OS rate of 64%. Finally, in the COG 99701 study, the use of VCR and carboplatin simultaneously in adjuvant treatment with cisplatin after radiotherapy yielded a 5-year OS rate of 81%. Meanwhile, high-dose myeloablative chemotherapy (HDC) and autologous stem cell rescue (ASCR) pre-protocols have been developed, showing interesting control rates. In 2003, Gururangan and colleagues reported a 4-year OS rate of 71% using an HDC regimen consisting of melphalan and cyclophosphamide or busulfan. In 2009, Chintagumpala and colleagues reported a 5-year OS rate of 67% after HDC regimens consisting of cisplatin, VCR, and cyclophosphamide. Finally, the SJMB03 trial evaluated a risk-adapted radiotherapy regimen followed by HDC, including cisplatin, VCR, and cyclophosphamide, followed by ASCR, with a reported 5-year OS rate of 100% in the high-risk group (metastatic and/or large residual disease) being 60%.At recurrence, HDC plus ASCR may be an interesting strategy. An HDC regimen including carboplatin, etoposide, and thiotepa before re-irradiation has also been proposed.6.2. Germ Cell TumorsLike its systemic counterpart, the chemosensitivity of pineal GCT (germ cell tumors) is well known. Conventional regimens are based on cisplatin or carboplatin and may include etoposide, cyclophosphamide, or ifosfamide. Pure germ cell tumors are highly sensitive tumors with favorable prognosis. The treatment for localized pure germ cell tumors consists of platinum-based chemotherapy plus radiotherapy. In patients with central nervous system metastasis, pure germ cell tumors can be cured with craniospinal irradiation without the need for additional chemotherapy. In the SIOP CNS GCT 96 study, 65 germ cell tumor patients received alternating treatment with carboplatin and VP16 prior to radiotherapy, with the “PE/IE” regimen. The 5-year PFS rate for this regimen was 88%, and the 5-year OS rate was 96%. In 2021, Bartels and colleagues reported results from a COG study evaluating the outcomes of using carboplatin and VP16 in reduced radiotherapy regimens after complete remission following chemotherapy. Although this trial did not demonstrate the non-inferiority of this approach, it was associated with an interesting chemotherapy response rate. Finally, the SIOP CNS GCT II trial evaluated the “PE/IE” combination treatment for non-metastatic germ cell tumors, followed by appropriate irradiation based on response quality. This very promising trial has now stopped enrollment, with results pending.Regimens containing platinum derivatives are also the platform for systemic treatment of non-germinomatous GCTs. The ACNS0122 and ACNS1123 protocols evaluated the regimens of carboplatin and etoposide with ifosfamide and etoposide alternated, yielding very encouraging results, showing a 5-year PFS rate of 84% and a 5-year OS rate of 93%. The SIOP CNS GCT 96 trial included 149 patients with non-germinomatous tumors, all receiving cisplatin, etoposide, and ifosfamide chemotherapy (PEI) followed by radiotherapy. This regimen was associated with a 5-year OS rate of 82% for localized patients and 75% for metastatic patients. Finally, the SIO CNS GCT II trial evaluated two chemotherapy regimens based on patient risk assessment, including PEI HDC combined with ASCR. Results are still pending, but this regimen may become a new first-line treatment for non-germinomatous GCT patients.At recurrence, GCT remains sensitive to chemotherapy. HDC regimens based on thiotepa or melphalan may be proposed, followed by ASCR. For non-germinomatous GCTs, the GEMPOX regimen consisting of gemcitabine, paclitaxel, and oxaliplatin has been associated with interesting responses in three patients.7. Future of Systemic TherapyDespite the current limited clinical research on the treatment of pineoblastoma or pineal parenchymal tumors of intermediate differentiation, there is increasing biological understanding of the various molecular subtypes of these diseases. In-depth studies of characteristic mutations in these tumors are expected to drive the development of targeted therapies. Meanwhile, optimization of chemotherapy regimens is being studied in pediatric settings, potentially leading to improvements in outcomes and side effects.