Original Authors: Rudelius M, Weinberg OK, Niemeyer CM, Shimamura A, Calvo KR.
Background/Introduction
ICC classifies pediatric myelodysplastic syndromes (MDS) separately from adult MDS. This separation acknowledges the unique biological characteristics of pediatric and adolescent MDS, which differ significantly from those of elderly MDS in terms of genetic landscape and treatment approaches. Pediatric MDS lacks recurrent mutations in epigenetic regulators or RNA splicing genes, which are known to expand clonal hematopoiesis in adults; instead, somatic alterations in mutations of SETBP1, ASXL1, RUNX1, and RAS/MAPK pathway define the genetic landscape.
Recently, new germline mutations in genes such as GATA2, SAMD9, or SAMD9L have been identified, which predispose to MDS at a young age. When primitive cells are not excessive, the histopathology of these cases typically shows a characteristic pattern defined as pediatric refractory cytopenia (RCC). The morphology of RCC is independent of the presence of clonal markers such as chromosomal abnormalities or somatic oncogenic mutations and may also be observed in some hereditary bone marrow failure syndromes (e.g., Fanconi anemia). ICC has updated the diagnostic criteria for RCC (Table 3) and recognizes that cases in this entity category may progress to MDS with excess primitive cells and develop into clonal hematopoiesis or severe bone marrow failure. As adaptive and maladaptive compensatory mechanisms are increasingly recognized, this evolutionary concept aligns with the challenges of bone marrow plasticity in young individuals. Furthermore, it allows for the adoption of risk-adapted treatment strategies and paves the way for future research.
Table 3 ICC Diagnostic Criteria for Pediatric Refractory Cytopenia (RCC)
|
|
||||
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Treatment options and prognosis for myeloid tumors vary by age. Compared to elderly MDS, treatment strategies for pediatric and adolescent MDS are typically curative. Especially for MDS with underlying germline susceptibility, early hematopoietic stem cell transplantation is the standard treatment, rather than intensified AML therapy. Therefore, the distinction between MDS and de novo AML is critical in pediatric myeloid tumors, ICC retains the definition of pediatric MDS-EB for MDS with 5-19% primitive cells, unlike adults, where 10-19% primitive cells replace MDS with MDS/AML.
Pediatric Refractory Cytopenia (RCC)
Definition
RCC is characterized by persistent cytopenia and at least two hematopoietic series or one series with ≥10% of cells showing dysplastic changes (Table 3). The histomorphological pattern defines RCC, with approximately 80% of cases exhibiting hypoplastic bone marrow. There is no excess of primitive cells, with primitive cells in bone marrow <5% and peripheral blood primitive cells <2%. While some RCC cases are true MDS, others may lack clonal evidence.
Clinical Manifestations
Patients present with symptoms of pancytopenia, such as bleeding, malaise, and infection, or may be asymptomatic. Lymphadenopathy secondary to local or systemic infections may occur, but hepatosplenomegaly is usually not a clinical feature of RCC. Other organ system abnormalities in patients with germline diseases may be due to underlying susceptibility. Compared to adult MDS, isolated anemia is rare in pediatric low-grade MDS, while most RCC patients exhibit relative age-appropriate erythrocytosis. Thrombocytopenia is most common, with severe neutropenia occurring in up to 25% of cases.
Morphological Features
Peripheral blood smears show macrocytic and anisocytotic dysplastic erythrocytes. Thrombocytopenia is usually accompanied by anisocytosis and few giant platelets. Neutrophils show dysplastic changes, such as pseudo Pelger-Huët anomaly, hypogranularity, and giant band forms, but dysplasia may be missed in severe neutropenia cases. There is no increase in primitive cells.
In bone marrow smears, erythroid hematopoiesis is characterized by megaloblastic changes, nuclear budding, or multinucleation. Granulocyte hematopoiesis shows nuclear-cytoplasmic maturation defects with hypogranularity and pseudo Pelger-Huët cells. Megakaryocyte generation may show aplastic features if present, characterized by small round or discrete nuclei. Micro megakaryocytes may be seen in some cases, supporting the diagnosis of RCC.
Most patients present with bone marrow hypoplasia; therefore, high-quality representative bone marrow biopsies are crucial for diagnosis. A hallmark of RCC is the patchy distribution of hematopoiesis (Figure 3A, E), which may not be apparent in bone marrow smears. The structure of the bone marrow is primarily disturbed by erythroid hematopoiesis, while granulocyte and megakaryocyte hematopoiesis is significantly reduced. Erythroid hematopoietic islands enlarge with dysplastic features and impaired maturation (Figure 3D). Granulocyte hematopoiesis is severely reduced and shows left shift. Megakaryocytes may be absent, with micro megakaryocytes rarely detected. Primitive granulocytes do not increase (primitive cells <5%). Hypercellular or normal RCC features similar morphological changes; dysplastic megakaryocytes require ≥10% of megakaryocytes with significant dysplastic features.(Table 3).
Identification of micro megakaryocytes requires immunohistochemical staining for megakaryocyte antigens such as CD61 or CD41 (Figure 3F), which is crucial for the differential diagnosis of severe aplastic anemia. CD34 immunohistochemistry aids in the quantification of primitive cells; however, it is important to exclude increased primitive cells stained with TdT and CD79a or PAX5.
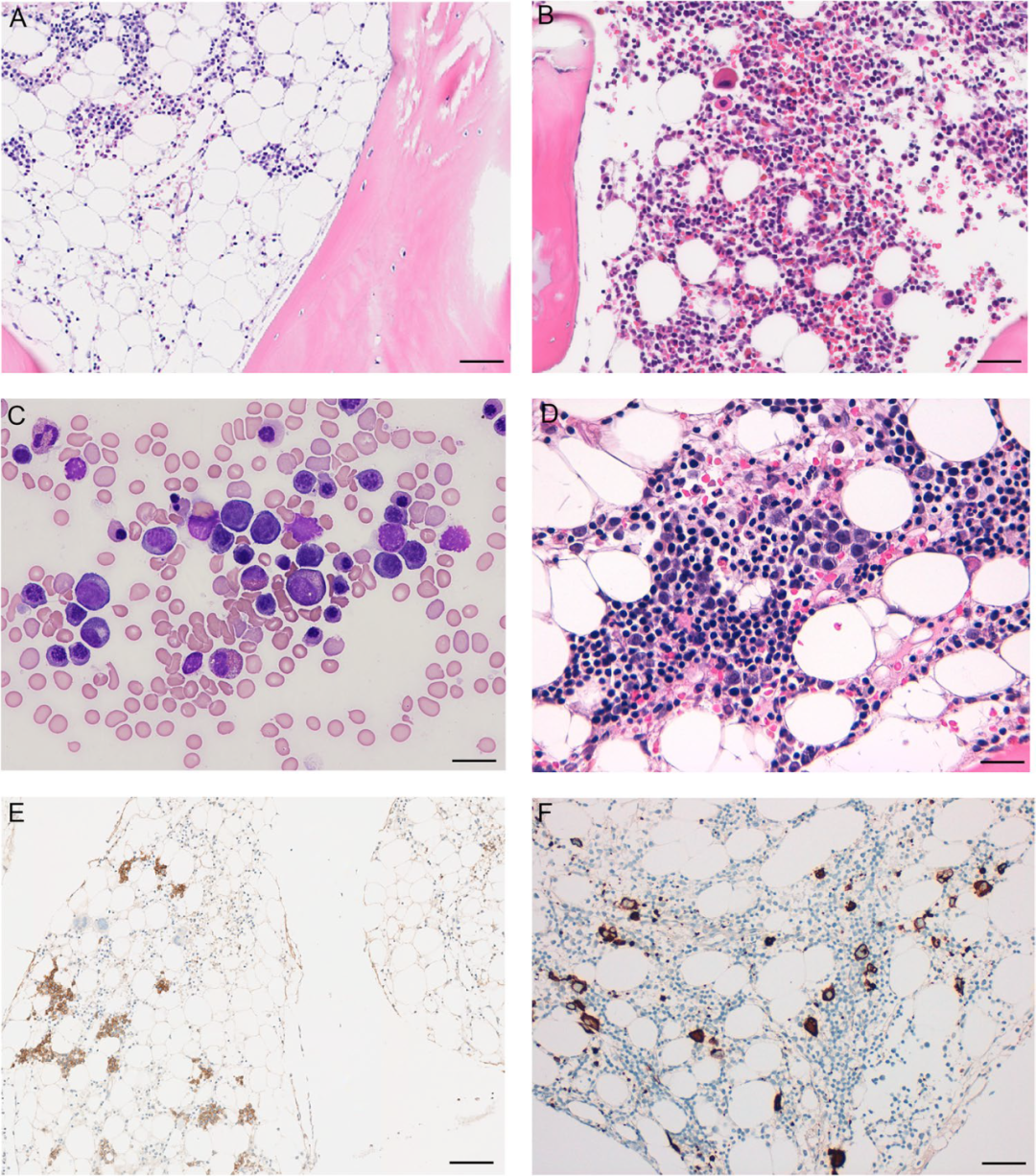
Figure 3 Pediatric Refractory Cytopenia. Hypoplastic RCC(A) and Normocellular RCC(B) H&E staining shows patchy distribution of erythroid hematopoiesis. E-cadherin (E) highlights the patchy distribution of erythroid hematopoiesis. Bone marrow smear (C) and bone marrow biopsy (D) show erythroid dysplasia (nuclear budding and maturation defects). CD61 immunohistochemical staining shows leafless megakaryocytes with few micro megakaryocytes(F. Scale bar:A, B, E, F 100µm; C 10µm; D 50µm
Cytogenetics and Molecular Genetics
Most RCC patients have normal karyotypes of hematopoietic cells. 7 Monosomy 7 is the most common cytogenetic abnormality, present in approximately 20% of RCC patients. GATA2 defects and SAMD9/SAMD9L germline diseases account for about half of these 7 monosomy cases. 8 Trisomy is the second most common chromosomal abnormality, with occasional random chromosome changes and complex karyotypes.
Somatic mutations in epigenetic regulators or RNA splicing genes are rare, while mutations in SETBP1, ASXL1, RUNX1, and RAS/MAPK pathway are the most common somatic abnormalities. Approximately 20% of RCC cases exhibit potential germline susceptibility, such as GATA2 defects, SAMD9/SAMD9L syndrome, Fanconi anemia, or RUNX1 defects.
Differential Diagnosis
The diagnosis of hypoplastic RCC is challenging and requires integration of clinical course, cytogenetics, molecular genetics, and morphology for accurate diagnosis. Various reactive bone marrow changes caused by infections, metabolic disorders (such as vitamin B12 deficiency), rheumatologic diseases, paroxysmal nocturnal hemoglobinuria, and autoimmune lymphoproliferative diseases may present with features similar to RCC. For the differential diagnosis of aplastic anemia, it is important to carefully assess dysplastic changes in all three series of blood cells. In this case, detecting micro megakaryocytes is particularly helpful for the diagnosis of RCC.
Myelodysplastic Syndromes (NOS)
Pediatric MDS cases without excess primitive cells and lacking RCC histomorphological patterns are classified as MDS not otherwise specified (MDS-NOS) (Table 4). This category also includes cases with monosomy 7 and del(7q) without cytopenia and/or dysplastic hematopoiesis. In rare cases, adult-type MDS, such as those with SF3B1 mutations or del(5q) tumors, may be seen in pediatrics; these cases can be classified according to adult MDS classification.
Table 4 Pediatric Refractory Cytopenia and Pediatric Myelodysplastic Syndromes
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
* Down syndrome and germline ETV6 or TP53 mutations are also predisposed to acute lymphoblastic leukemia
** Excludes defining AML cytogenetic abnormalities
*** If cytogenetic abnormalities defined by MDS are present, cases without cytopenia are included
MDS with Excess Primitive Cells
Pediatric MDS with excess primitive cells is defined as MDS with 5-19% bone marrow primitive cells or 2-19% peripheral blood primitive cells (Figure 4). In pediatric patients, the genetic abnormalities or clinical course with more fixed thresholds for primitive cells better define MDS and AML. Additionally, diseases with defined cytogenetic abnormalities of NPM1 or biallelic CEBPA mutations at any age are considered AML, regardless of the percentage of primitive cells. However, treatment options and prognosis for myeloid tumors vary by age. Most pediatric MDS-EB patients receive allogeneic hematopoietic stem cell transplantation without prior AML induction chemotherapy; thus, the distinction between de novo AML and MDS is critical for improving cure rates in young patients with myeloid tumors.

Figure 4 Myelodysplastic Syndromes with Excess Primitive Cells. Bone marrow biopsy H&E staining shows hyperplastic hematopoiesis with structural destruction, granulocyte maturation defects, and mononuclear megakaryocytes(A). Primitive granulocytes highlighted by CD34 immunohistochemical staining(B). Scale bar:A, B 50µm.