–01–
Introduction
The announcement of the 2022 Nobel Prize in Chemistry brought the cutting-edge technology of “Click Chemistry” into the public eye. The groundbreaking work of three scientists has not only revolutionized the field of organic synthesis but also opened new pathways for cancer treatment—specifically, the development of Antibody-Drug Conjugates (ADC) is advancing towards a new era of higher precision and lower toxicity, empowered by click chemistry.
–02–
What is Click Chemistry?
Click chemistry is an efficient and precise molecular synthesis technique, based on the core idea of rapidly and selectively connecting molecular modules through chemical reactions, akin to “building with Legos”. This technology was proposed by Nobel Laureate Barry Sharpless in 2001 and has the following core characteristics:
1. Efficiency: Reactions can be completed quickly under mild conditions, with yields exceeding 90% and minimal by-products.
2. Modular: Flexible assembly of different molecular modules is achieved through standardized reactive groups (such as azides and alkynes).
3. Bioorthogonality: Reactions can safely occur in living environments (such as cells and blood) with minimal interference with normal physiological processes.
–03–
Three Core Advantages of Click Chemistry Empowering ADCs
1. Precise Targeting: Goodbye to the “Random Coupling” Era
Early ADCs coupled drugs randomly through the lysine or cysteine residues of the antibodies, leading to uneven drug-antibody ratios (DAR) and significant variability in efficacy.Click chemistry introduces specific functional groups (such as azides and alkynes) on the antibody and drug, achieving a “one-to-one” precise connection, with controllable DAR values exceeding 96%.
2. Superb Stability: Molecular-Level “Rivet Technology”
Click chemistry reactions (such as CuAAC, SPAAC) form cyclic structures (such as triazole rings) that exhibit chemical inertness, significantly enhancing acid and enzyme resistance. Experimental data shows that ADCs constructed based on SPAAC technology maintain over 95% integrity after being stored in serum for 3 days, far superior to traditional thioether bonds.
3. Diverse Application Scenarios: “Modular” Drug Design
By employing a “click-release” strategy, drugs can be activated in a targeted manner. For example, the bispecific ADC M1231 uses the IEDDA reaction to release toxic payloads only in tumors co-expressing HER2/HER3. Bispecific ADCs utilize branched linkers to carry drugs with different mechanisms (such as microtubule inhibitors + DNA damaging agents), achieving dual anti-tumor effects through a single click reaction.
–04–
Practical Applications of Click Chemistry in ADC Synthesis
The application of click chemistry in the synthesis of ADCs mainly includes the following reactions:
Copper-Catalyzed Azide-Alkyne Cycloaddition (CuAAC)
CuAAC is the core reaction of click chemistry, utilizing Cu(I) to catalyze the reaction between azides and alkynes to generate 1,4-disubstituted triazole bonds, offering advantages such as high yield, high selectivity, and mild conditions. In ADCs, CuAAC is commonly used for the conjugation of antibodies with linkers or cytotoxic agents, for example, by coupling azide-labeled antibodies with alkyne-modified drug molecules. However, the cytotoxicity of copper ions limits its application in living systems.
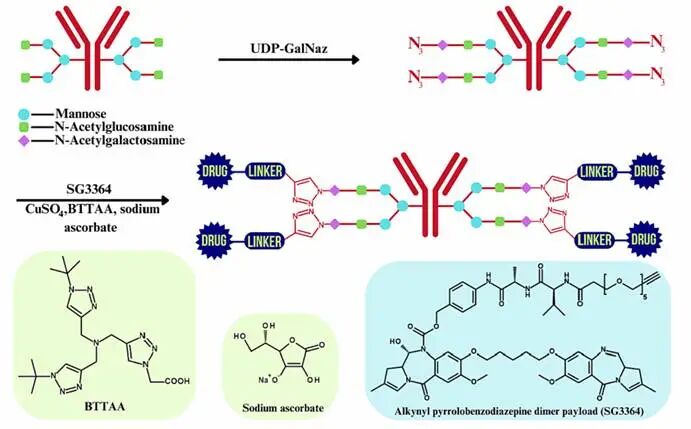
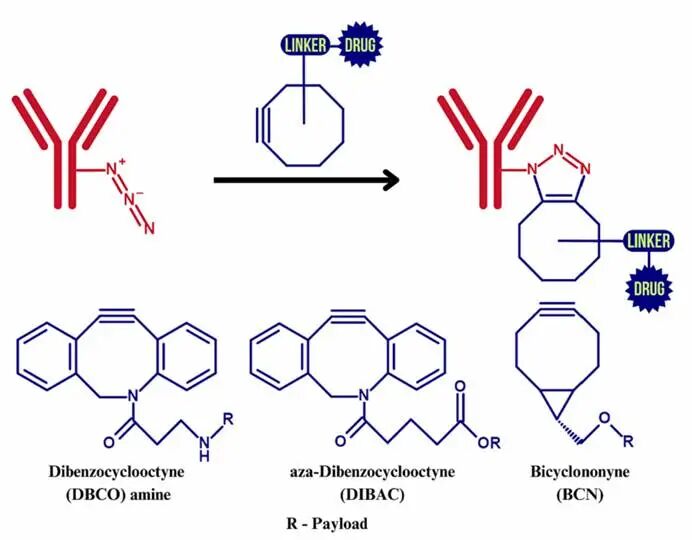
Strain-Promoted Azide-Alkyne Cycloaddition (SPAAC)
SPAAC lowers the activation energy through the ring strain of cyclooctyne, allowing for efficient reactions without metal catalysts, thus addressing the biocompatibility issues of CuAAC. Studies have successfully constructed high DAR (Drug-Antibody Ratio) ADCs targeting leukemia by coupling LILRB4 antibodies with MMAF using SPAAC.
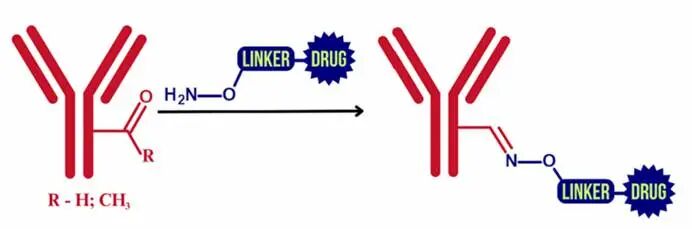
Oxime Ligation
Utilizing the condensation reaction of aldehydes/ketones with aminooxy groups, the conjugation sites can be controlled. For example, studies have shown that coupling the aldehyde-modified antibodies with aminooxy-functionalized drugs can form oxime bonds. Although early oxime bonds were limited in application due to acid-base sensitivity, optimized systems have shown improved serum stability.
 Example
Example
Michael Addition
The Michael addition reaction is one of the most core chemical coupling strategies in ADC synthesis, especially dominating in cysteine-based random coupling techniques. This reaction achieves efficient coupling of antibodies with cytotoxic agents through the conjugate addition of α,β-unsaturated carbonyl compounds (such as maleimides) with thiols.

Hydrazone-Pyrrolidine-Spiegler Linkage (HIPS)
Stable C-C bonds are formed through the condensation of aldehydes with hydrazones. For example, using formylglycine to generate enzymes that introduce aldehyde groups into antibodies, followed by reaction with drug linkers containing pyrrolidine, specific ADCs can be generated. Experiments show that the in vitro activity of this linkage method, HIPS-ADC, is comparable to approved ADCs and inhibits tumor growth in animal models.

Diels-Alder Reaction (DA)
Forward DA reaction: Achieves stable coupling under mild conditions through the cycloaddition of diene (such as cyclopentadiene) with dienophiles (such as maleimides). Studies have proven that ADCs prepared through this reaction exhibit better serum stability than traditional thiol-maleimide linkages.
Inverse Electron Demand DA (IEDDA): Rapid reactions between tetrazines and cyclooctene compounds (rates up to 10³–10⁶ M⁻¹s⁻¹) are suitable for living environments. For example, antibodies carrying cyclooctene can connect with tetrazine-modified drug MMAE through IEDDA, significantly enhancing coupling efficiency and targeting.

–05–
Breakthroughs and Challenges: The Unfinished Journey of Click Chemistry
1. Optimization of In Vivo Reaction Efficiency Although in vitro experiments show high efficiency of click reactions, complex factors in living environments (such as blood flow and osmotic pressure) may reduce coupling efficiency. Solutions include developing more reactive cyclooctyne derivatives (such as DIBAC).
2. Balancing Linker Stability Overly stable linkers may prevent drug release, while overly reactive linkers may decompose in blood. New “dual-responsive” linkers (such as those responding to both pH and enzymes) are currently being validated in clinical trials.
3. Cost Control in Scalable Production Copper-free click reagents (such as DBCO) are expensive, limiting industrial applications. Advances in synthetic biology techniques (such as microbial fermentation production of cyclooctyne) are expected to reduce costs.
–06–
Conclusion
From the laboratory to the clinic, click chemistry is driving ADCs from a “broad-spectrum” to a “precision” approach. Currently, over 20 click chemistry-derived ADCs have entered clinical trials, covering indications such as breast cancer, lymphoma, and solid tumors. In the future, with the iteration of bioorthogonal tools and interdisciplinary collaborations (such as AI-assisted linker design), click chemistry is expected to fundamentally rewrite the landscape of cancer treatment, providing patients with higher quality of life and longer survival times.
References:
1. Click chemistry in the synthesis of antibody-drug conjugates. Bioorg Chem. 2024 Feb:143:106982.
Reply “ADC” in the public account or scan the QR code in the image below to download the PDF version of the e-book “Antibody-Drug Conjugates: From Basics to Clinic” for free!

The public account has established a “Professional Exchange Group for Pharmaceutical Discussions” WeChat industry group and reader group. Scan the editor’s QR code below to join. Please inform your name, workplace, and position when joining the industry group..
