Nanosuspensions (NS) are considered dispersions of drug particles with a particle size of less than 1μm (most commonly 200-500nm) that are stabilized by adding suitable polymers and/or surfactants. By dispersing drugs into nanoscale particles, nanosuspensions significantly increase the specific surface area of the drug, thereby promoting drug dissolution and significantly improving its release performance and bioavailability. However, stability issues severely limit the widespread application of this technology in drug delivery systems. Nanoparticles, due to their high surface energy, are prone to aggregation, sedimentation, or crystal growth problems. This can lead to a loss of their original advantages, affecting their dissolution performance, stability, and efficacy. Therefore, stabilizers are usually required to prevent particle aggregation and ensure system stability. Stabilizers typically adsorb onto the surface of nanoparticles to ensure the stability of the nanosuspension through a combination of electrostatic repulsion and steric hindrance. This adsorption and stabilization mechanism appears to be highly specific, but is poorly understood and difficult to predict. Current stabilizer screening largely relies on empirical methods, often involving extensive experimental screening to determine formulations of nanosuspensions with sufficient stability. However, this process is time-consuming, labor-intensive, and costly, making it very unfriendly for drug development. Especially for newly discovered compounds, traditional experimental screening methods are often limited by material scarcity and cost, which affects further research and development.
To address the above issues, Tianjin University of Traditional Chinese MedicineResearcher Li Wenlong and his research group established a connection between microscopic simulations (molecular self-assembly studies of different stabilizers with drugs) and macroscopic experiments (key quality attributes of NS) based on computer-aided drug design methods, aiming to create a new pathway for rational formulation design. The relevant research results were published in the International Journal of Pharmaceutics, with master’s student Zhang Xiaoyang as the first author of the paper.
Table 1 shows the stabilizer screening results obtained from three different methods: real experiments, molecular docking (MD), and molecular self-assembly. Based on the MD results (Figure 1), PVP K30 is the best stabilizer for Tet-NS. However, there is a significant discrepancy between this result and the real experimental results, which may be attributed to several issues. First, during the actual preparation of NS, the stabilizer and drug molecules are not in a static state. The interactions between the stabilizer and the drug are not a simple ‘one-time’ binding, but are constantly adjusting and changing in solution. MD does not take these dynamic processes into account, and therefore may not accurately reflect the actual interactions between the drug and stabilizer at the nanoscale. Second, in NS, drug molecules and stabilizers often have multiple intermolecular interactions. For example, multiple stabilizer molecules may bind to drug molecules simultaneously, forming a multi-molecular binding state. However, MD typically simulates only a single binding mode and does not consider these complex many-body effects. Third, the binding energy values provided by MD are based on thermodynamic calculations, meaning they indicate the optimal binding state that stabilizers and drug molecules may form under specific conditions. However, the stability of NS depends not only on thermodynamic stability but also on the kinetic behavior of the system, such as particle collisions and aggregation. These kinetic effects are often not fully represented in static MD.
Table 1 Stabilizer screening results obtained from different methods
|
Method |
Stabilizer Screening Results |
|
Real Experiment |
PVA>Span60>SDS>PVP K30>PEG4000>P188 |
|
Molecular Docking |
PEG 4000>PVA>SDS>P188>Span60>PVP K30 |
|
Molecular Self-Assembly |
PVA>Span60≥SDS>PVP K30≥PEG4000≥P188 |
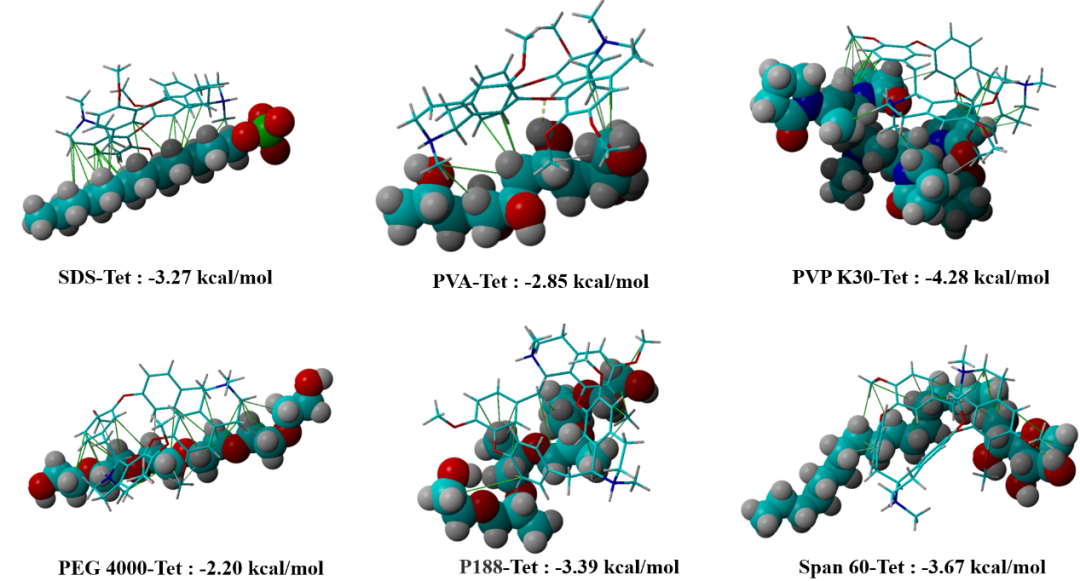
Figure 1 Molecular docking results of drug and stabilizer molecules (green solid lines represent hydrophobic forces, yellow dashed lines represent hydrogen bonds)
The virtual screening results based on the molecular self-assembly strategy (Figure 2) are almost consistent with the real experimental results (Figure 3). Compared to the static binding MD technique, the molecular self-assembly technology based on molecular dynamics simulation can more accurately screen suitable stabilizers for NS. This is because MDS can more realistically simulate the nanoparticle formation process. MDS considers the dynamic behavior of molecules, including conformational changes, solvent effects, and molecular motion, thus allowing for the evaluation of the binding modes and stability between stabilizers and drug molecules in a state closer to the real environment. This simulation can capture not only the instantaneous interactions between stabilizer molecules and drugs but also observe their long-term evolution in solution, further predicting which stabilizers can effectively bind and optimize drug stability. Additionally, MDS can assess SASA, Rg, RMSD, and how the interactions between stabilizers and drug molecules change over time, providing a more comprehensive and dynamic perspective for selecting the most suitable stabilizer.
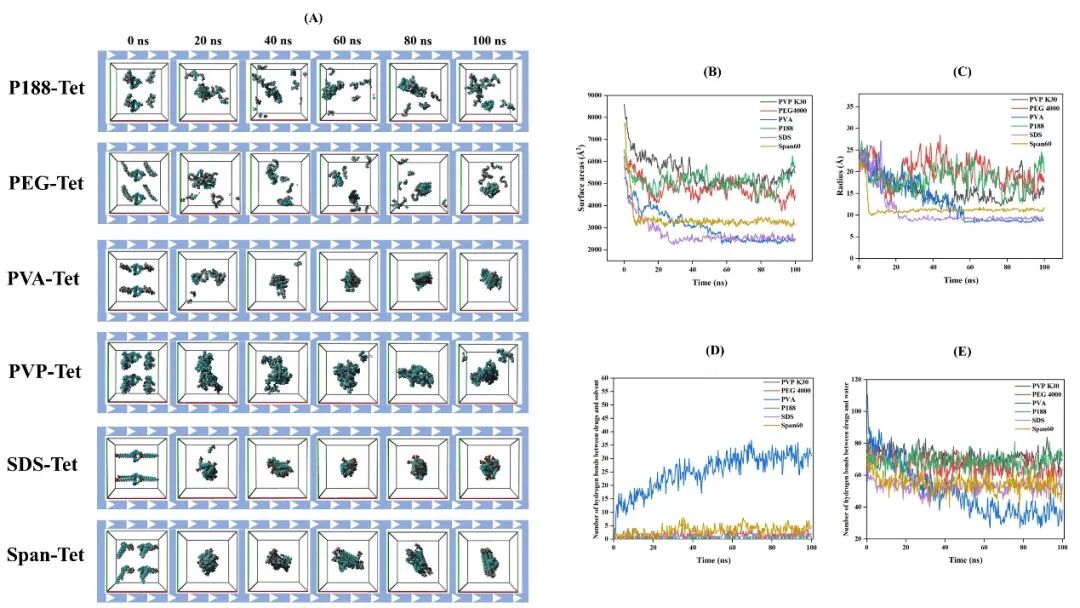
Figure 2 (A) Simulation trajectories of different stabilizer systems; (B) Solvent-accessible surface area of different stabilizer systems; (C) Radius of gyration of different stabilizer systems; (D) Hydrogen bonds between drug and stabilizer; (E) Hydrogen bonds between drug and water
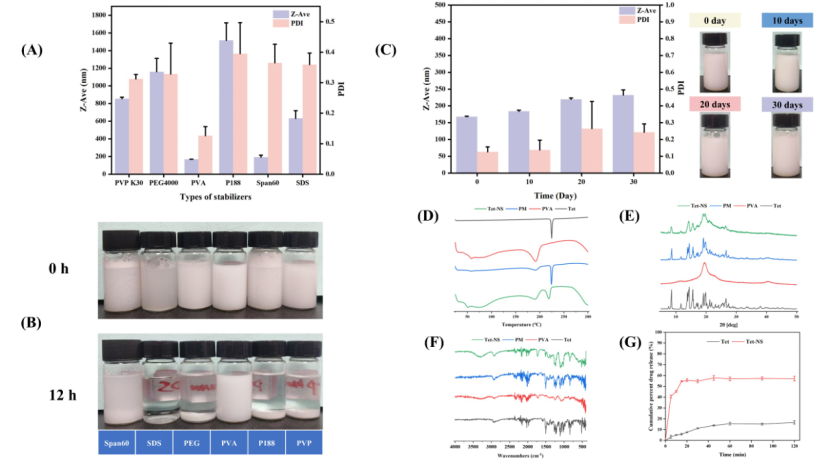
Figure 3 (A) Z-Ave and PDI of nanosuspensions prepared with different stabilizers; (B) Appearance of nanosuspensions prepared with different stabilizers within 12 hours; (C) Short-term stability test results; (D) DSC results of Tet-NS, PM, Tet, PVA; (E) XRD results of Tet-NS, PM, Tet, PVA; (F) FT-IR results of Tet-NS, PM, Tet, PVA; (G) In vitro dissolution test results
This strategy can significantly reduce the candidate range of stabilizers through virtual screening, theoretically greatly shortening the formulation development cycle and reducing R&D costs. Furthermore, microscopic simulations can provide important insights into how excipient-drug interactions determine the stability of NS.
References:Xiaoyang Zhang, Wenlong Li, et al. A new strategy for nanosuspension stabilizer screening based on computer-aided drug design and molecular self-assembly. International Journal of Pharmaceutics. 2025, DOI:10.1016/j.ijpharm.2025.125860.