Abstract Depression is a mental disorder characterized by significant and persistent low mood. Recent studies have found that cholecystokinin (CCK), a small molecule brain-gut peptide (BGP), is widely distributed in the central nervous system (CNS) and the enteric nervous system (ENS). To clarify the relationship between CCK and the pathogenesis of depression, this article reviews the role and mechanisms of CCK in depression, including the hypothalamic-pituitary-adrenal axis (HPA), synaptic function, molecular and circuit mechanisms.
Keywords Depression; Cholecystokinin; Neurons; Synapses
Depression is a global neuropsychiatric disorder that severely affects quality of life, with main clinical symptoms including persistent low mood, decreased self-esteem, aversion to activities, sleep or appetite disturbances, and difficulty concentrating[1]. Over 300 million people of all ages worldwide suffer from depression, making it one of the leading causes of loss of productivity[2]. Cholecystokinin (CCK), a typical brain-gut peptide secreted by intestinal I cells and neurons after food intake, is widely distributed in the gastrointestinal tract (duodenum, jejunum, and colon) and the central and peripheral nervous systems, playing an important physiological role as a neurotransmitter. CCK not only plays a significant role in gastrointestinal function regulation but also has an important role in emotional regulation within the central nervous system[3]. This article reviews the role and mechanisms of CCK in depression, providing new clues and ideas for future research on the pathogenesis of depression and the development of antidepressants.
1 CCK

CCK is a known digestive hormone and the most abundant peptide neurotransmitter in the brain, involved in various functions including stress regulation, anxiety, and panic attacks; it is widely distributed in the central and peripheral nervous systems, particularly in the cerebral cortex, amygdala, hippocampus, pituitary, and the solitary nucleus of the medulla. CCK coexists with neurotransmitters such as dopamine and oxytocin in nerve cells and regulates other neurotransmitters like γ-aminobutyric acid (GABA), glutamate, dopamine, and acetylcholine. However, further research is needed to determine whether the regulatory effects of CCK on these neurotransmitters are related to the occurrence and development of depression[4].
CCK receptors belong to the family of G-protein-coupled receptors (GPCRs) and have seven transmembrane helices. CCK receptors are widely distributed in the brain, such as in the cortex, hippocampus, and amygdala. There are two types of CCK receptors: CCK1R and CCK2R. CCK exerts its effects through these two receptor subtypes, having significant physiological and pharmacological effects involved in the pathogenesis of certain mental disorders such as memory, anxiety disorders, and schizophrenia[5]. Upon binding to CCK receptors, CCK activates phospholipase C (PLC), leading to increased intracellular inositol triphosphate (IP3) and Ca2+ levels, achieving transmembrane signal transduction[6]. CCK1R is activated by sulfated CCK (CCKs) and is released in the gastrointestinal tract and mammalian brain, functioning similarly to gastrin, regulating appetite intake and inhibiting appetite. It is mainly distributed in peripheral nerves, including vagal afferent terminals in the mucosal epithelium, and is also found in the area postrema (AP), dorsomedial hypothalamus (DMN), solitary nucleus, and hypothalamus in the central system. CCK2R is primarily distributed in the cerebral cortex, substantia nigra, amygdala, olfactory bulb, lateral septal nucleus, striatum, hippocampus, hypothalamus, red nucleus, dorsal tegmental area, cerebellum, and vagus nerve in the central nervous system. CCK2R plays a major role in pain perception and opioid dependence, unrelated to CCK1R, and does not regulate food intake. Notably, in the cerebral cortex and limbic system, CCK exerts effects through CCK2R in stress and anxiety-related emotional behaviors. Behavioral studies with CCK receptor agonists and antagonists have shown that CCK receptor agonists can increase anxiety and depression-like behaviors, while CCK2R antagonists exhibit anxiolytic and antidepressant effects[8-9], although some researchers believe that the injection site of CCK antagonists is a major determinant of anxiety behavior.
CCK exists in various biologically active molecular forms, which are processed from the CCK precursor mRNA sequence containing 115 amino acid residues. The main molecular forms include CCK8, CCK4, CCK5, CCK7, CCK33, CCK39, and CCK58; the forms most related to depression are CCK8 and CCK4[10].
CCK8 is the most abundant form in the body and the primary form of CCK in brain neurons, present in almost all CCK molecules, serving as the basic unit of CCK’s peripheral and central biological activity. The sulfated CCK8 (CCK8S) has the strongest biological activity. In the central nervous system, CCK primarily exerts its biological effects through CCK8S. CCK8S is involved in many important physiological and pathophysiological processes, such as behavior, anxiety, learning/memory processes, and neurogenic pain. CCK8 acts on target cells through various mechanisms, including endocrine, paracrine, and autocrine, binding to CCK1R or CCK2R, mediating corresponding adaptive cell protective effects[11], with CCK8 showing significantly higher affinity for CCK2R than for CCK1R. CCK8 directly acts on T cells, exerting anti-inflammatory and immune regulatory effects[12], and can significantly inhibit the increase of TNF-α, IL-1β, and IL-6 induced by lipopolysaccharide (LPS) in rats[13]. CCK8 is the most abundant in the central nervous system and is currently known as the strongest endogenous anti-opioid peptide, significantly inhibiting the formation of conditioned place aversion (CPA) induced by chronic morphine dependence and naloxone-precipitated withdrawal. An acute toxicity experiment in rats showed that acute morphine intervention has a significant anxiolytic effect, while CCK8 has an anxiogenic effect. However, exogenous CCK8 can significantly inhibit the anxiety-like behavior induced by spontaneous withdrawal from morphine in rats by upregulating endogenous opioid peptides through CCK1R[14].
CCK4 is another form of CCK, expressed abundantly in the cerebral cortex and limbic structures, selectively exerting effects through CCK2R, particularly well-known for its role in emotional regulation. Some preclinical and clinical studies have shown that administration of CCK4 can induce panic and anxiety responses[15]. For example, in a double-blind, placebo-controlled study, 26 out of 30 subjects exhibited significant panic responses upon receiving CCK4 stimulation[16].
2 The Role of CCK in Depression
Research both domestically and internationally has found multiple single nucleotide polymorphism (SNP) loci in the CCK gene, among which the -45C/T and -196G/A variants are correlated with various mental disorders such as depression, suicidal behavior, and Parkinson’s disease[17]. The genetic polymorphisms of the CCK receptor gene are closely related to abnormal behaviors such as alcoholism, personality disorders, thought disorders, anxiety, and fear. The human CCK1R gene is located at 4p15.1~15.2, adjacent to the D5 receptor gene (4p15.1~15.3), consisting of 5 exons and 4 introns, with a length of 21.8 kb. Studies have found that the allele frequency of the CCK1R gene promoter site -286 and the AA genotype frequency, as well as the -333 site GG genotype frequency, are significantly higher in schizophrenia patients than in the control population[6]. Research indicates that the frequency of the T allele at the CCK1R gene rs1800857 locus is significantly higher in the schizophrenia group than in the healthy control group, suggesting that the T allele at this locus is a risk factor for schizophrenia, making individuals carrying the T allele more susceptible to schizophrenia[18]. The human CCK2R gene is located at 11p15.4, adjacent to the DA and D4 receptor genes. This gene consists of 5 exons and 4 introns, with a length of 11 kb. In suicidal populations, the expression of the CCK2R gene is significantly enhanced in the cerebellum, cingulate gyrus, and frontal cortex, but no correlation has been found between the polymorphisms of the CCK2R gene and mental disorders such as schizophrenia.
Early clinical studies have found that CCK levels in the cerebrospinal fluid of depressed patients are negatively correlated with certain depression parameters, and patients who have attempted suicide once or multiple times often have higher CCK levels than those who have not attempted suicide; however, it was further found that CCK levels in cerebrospinal fluid are not correlated with 5-hydroxyindoleacetic acid (5-HIAA) and plasma cortisol[19]. Tove et al.[20] found that depressive symptoms in patients with major depression are negatively correlated with CCK4 levels in cerebrospinal fluid. The current research on CCK levels in the cerebrospinal fluid of depressed patients remains controversial and requires further investigation. Researchers found that in a study of 25 suicide attempters and 23 healthy controls, using the Hamilton Depression Scale (HDRS) for scoring and measuring plasma CCK levels, the CCK levels in suicide attempters were 22.67 times higher than in healthy controls, and HDRS scores were 14.33 times higher than in healthy controls. However, the correlation between CCK levels and HDRS scores was minimal, indicating that CCK levels are unrelated to depression scores. Suicidal ideation, plans, and attempts have different etiologies, and various neuropsychiatric disorders and cognitive conditions may influence CCK levels[21]. Other potential but unassessed psychological variables, such as anxiety, stress, agitation, impulsivity, and fearlessness, may better describe the relationship between psychological function and CCK. Therefore, future research should carefully and comprehensively assess relevant influencing factors.
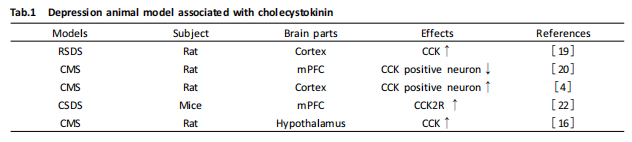
Existing literature reports that in the brains of rats subjected to stress or anxiety-like stress, CCK levels, CCK receptor density, and CCK release increase[22]. Kim et al.[23] found that in chronic mild stress (CMS) model rats, CCK expression significantly increased in brain regions including the paraventricular hypothalamic nucleus (PVN), periventricular hypothalamic nucleus (PE), paraventricular thalamic nucleus (PV), and arcuate nucleus (ACN). Studies have shown that increased CCK levels in the amygdala lead to CCK dysfunction, resulting in severe depression[24]. In a repeat social defeat stress (RSDS) experiment in rats, the release level of CCK in the cortex significantly increased[25]. Czéh et al.[26] found that 7 weeks of CMS stress can reduce the number of CCK-positive GABAergic neurons in the medial prefrontal cortex (mPFC) of male Wistar rats. After 7 weeks of CMS, the density of CCK-positive neurons in the ventral orbitofrontal cortex significantly increased in adult male rats[4]. Knockout of CCK in the basolateral amygdala (BLA) of mice has anxiolytic and antidepressant effects[24], and related studies indicate that overexpression of CCK in the BLA is one of the key signals for the development of depression. Researchers conducted conditioned fear experiments on normal rats and rats with CCK receptor gene knockdown in the BLA, finding that the percentage of immobility in rats with CCK2R knockdown in the BLA significantly decreased at 1 day and 28 days after foot shock, further confirming that knockdown of CCK2R in the BLA can impair the consolidation of fear memory[27]. Reduced neuronal activity in the mPFC of mice is associated with depression and anxiety-like behaviors induced by social defeat, and Vincent et al.[28] found that selective induction of the transcription factor ΔFosB overexpression in the mPFC of mice in a chronic social defeat stress (CSDS) model leads to reduced neuronal activity, and this effect of the transcription factor is partially mediated by inducing CCK2R, suggesting that reduced neuronal activity in the CCK2R pathway may be involved in the pathogenesis of depression.
Moreover, studies have indicated that the CCK system may be a relevant target for novel antidepressant treatments. Therefore, further understanding the mechanisms by which CCK affects depression plays an important role in the research process of depression, providing a theoretical basis for future elucidation of the pathogenesis of depression.
3 Mechanisms by Which CCK Affects Depression
3.1 Hypothalamic-Pituitary-Adrenal Axis (HPA)
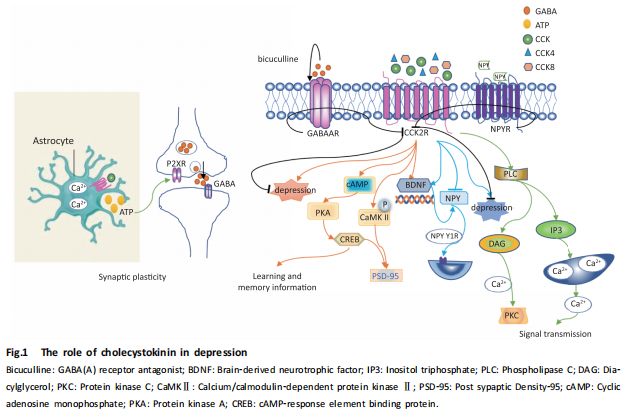
Disruption or insufficiency of HPA axis function can easily induce depression. Currently, it is commonly found that the secretion of corticotropin-releasing hormone (CRH), adrenocorticotropic hormone (ACTH), and cortisol is impaired in depression, with cortisol mediating changes in HPA axis negative feedback through mineralocorticoid receptors (MR) and glucocorticoid receptors (GR)[29]. However, drugs that directly regulate HPA function for the treatment of depression are still in the validation stage. CCK-expressing neurons are present in several regions involved in regulating HPA axis activity, particularly in the hypothalamus, but also in limbic regions. In mouse experiments, selective loss of tyrosine kinase receptor B (TrkB) signaling in CCK neurons induces glucocorticoid resistance, leading to increased CRH expression, chronic cortisol elevation, and dysregulation of HPA axis activity, resulting in metabolic disorders[30].
Intraperitoneal injection of CCK8 in rats can increase plasma cortisol levels, while in vitro experiments show that high doses stimulate the pituitary to secrete ACTH and promote the hypothalamus to secrete CRH, indicating that CCK acts on the entire HPA axis. Intravenous injection of CCK8 in rats shows a dose-dependent stimulation of ACTH release, which can be blocked by the CCK receptor antagonist (L-364,718). After severing the pituitary axis, the reactivity of ACTH and cortisol to CCK8 significantly decreases, indicating that exogenous CCK8 primarily acts on the HPA axis through CRH[31]. Peripheral injection of CCK4 can increase plasma ACTH and cortisol levels[32]. Researchers injected CCK4 intravenously into 30 healthy male subjects, which could trigger panic anxiety symptoms and increase ACTH and cortisol release acting on the HPA axis[33], and related studies have proven that CCK4-induced panic is associated with HPA axis activation[34]. Intravenous injection of CCK4 increases serum cortisol levels in MS depression model rats, indicating that CCK4 enhances the HPA axis response in MS stress rats[35]. In RSDS experiments in rats, stress causes an increase in CCK release in the cortex, and the specific antagonist of CCK2R, CI-988, can reduce the immobility time of rats in the FST experiment, preventing excessive activation of the HPA axis, reduction of hippocampal volume and cell proliferation, and decreased sweet intake, indicating that antagonizing CCK in the cortex of RSDS rats can regulate HPA axis function, while CCK1R antagonists do not show corresponding effects in depression models[24], and long-term treatment with the antidepressant imipramine can reverse the aforementioned depressive behaviors. Studies have shown that transgenic mice with overexpression of CCK2R exhibit hyperactivity of the HPA axis after acute stress[36].
In summary, CCK4 and CCK8 have significant stimulatory effects on the HPA axis, leading to their impact on emotions such as panic, anxiety, and depression, which can be reversed by selective CCK receptor antagonists.
3.2 Molecular Mechanisms
Brain-derived neurotrophic factor (BDNF), as an important neurotrophic factor, plays a positive role in neuronal plasticity and repair after injury, mediating the antidepressant effects of antidepressants. BDNF plays an important role in the pathogenesis and treatment of mental disorders, regulating neuropeptides such as NPY and CCK, which are related to mood disorders such as depression[37]. Serum BDNF levels in depressed patients are significantly lower than those in healthy controls[38]. Animal experiments have shown that in the forced swimming test, BDNF-deficient mice are resistant to antidepressants. The expression of pituitary adenylate cyclase-activating polypeptide (PACAP) gene in neurons can be synergistically increased by Ca2+ influx and intracellular cAMP signaling, participating in the regulation of synaptic plasticity and emotional regulation among various physiological functions; BDNF expression is regulated by PACAP, playing an important role in promoting neuronal survival, differentiation, and synaptic plasticity. Studies have found that increased PACAP gene expression synergistically elevates the expression of prodynorphin (PDYN), CCK, and preprotachykinin (PPT) mRNA in primary cultured rat cortical neurons, leading to increased BDNF mRNA expression through specific pathways[39], providing a theoretical basis for further exploring the occurrence and treatment of mental disorders.
Studies have found a statistically significant association between 12 SNPs of CCK2R and 5 SNPs of BDNF in suicide attempters, with 7 SNPs of CCK2R still associated after multiple testing corrections; however, no association was found between CCK and suicidal behavior[40]. It is speculated that certain polymorphisms of CCK2R may be potential triggers for suicidal behavior in bipolar disorder. Maron et al.[41] found that after injecting CCK4 into healthy subjects, those with stronger anxiety responses exhibited higher BDNF levels than those with mild anxiety. Nawa et al.[42] found that injecting BDNF into the ventricles of newborn rats increased the expression of neuropeptides such as NPY and CCK in the brains of newborn rats. Injection of CCK4 into the lateral ventricle of mice increased immobility time in the forced swimming test, and the depressive effect of CCK4 was enhanced by co-injection of NPY Y1 receptor antagonists and weakened by co-injection of NPY and NPY Y1 receptor agonists[43], suggesting that NPY and its receptors may mediate the depressive effects of CCK4. Kim et al.[23] found that after 8 weeks of CMS modeling, CCK synthesis increased in several subregions of the PVN, hypothalamic ventricle nucleus, thalamic periventricular nucleus, and arcuate nucleus, while NPY synthesis decreased, suggesting that NPY and CCK play opposing roles in regulating stress responses. These findings indicate that the CCK and NPY systems may play important roles in the pathophysiology of symptoms related to chronic stress responses, such as depression and anxiety-related disorders. Sakaida et al.[44] found that ECS treatment reduced CCK expression in the PVN of depression model mice, further confirming the close relationship between CCK and mental disorders such as depression. The relationship between CCK, CCK receptors, neurotrophic factors BDNF, and neuropeptide NPY is closely related to the occurrence and development of depression.
3.3 Synaptic Mechanisms
Studies have found that CCK neurons express high levels of cannabinoid receptors presynaptically, suggesting that CCK neurons play a role in synaptic plasticity changes related to learning and memory[45]. Existing studies have shown that CCK can enhance long-term potentiation (LTP) in the hippocampus and improve spatial memory in experimental animals. Research has found that CCK can regulate synaptic plasticity under different nutritional states; during fasting, ghrelin activates AMP-activated protein kinase (AMPK) through its receptors, inhibiting mechanistic target of rapamycin (mTOR) and blocking the induction of long-term depression (LTD); when food is reintroduced, vagal nerve excitation can activate CCK and act on the N-methyl-D-aspartate receptor (NMDAR)-extracellular regulated protein kinases (ERK) pathway, altering the balance between AMPK and mTOR, thereby facilitating the induction of LTD in the solitary nucleus[46]. Research using stereotaxic surgery, placing stimulating and recording electrodes in the perforant path and dentate gyrus of rats, showed that intraperitoneal injection of CCK8S had a transient excitatory effect on baseline responses but inhibited paired pulse indices during the acute phase; thus, it is believed that the short-term effects of CCK on synaptic function are time-dependent, exhibiting either stimulatory or inhibitory effects at different time points[47]. Malihe et al.[48] found that high levels of CCK8S during stress induction can regulate the damaging effects of stress on hippocampal synaptic plasticity and memory. In animal models of post-traumatic stress disorder (PTSD), high expression levels of CCK and its receptors were found, and knocking out CCK1R and CCK2R genes sequentially revealed that CCK2R in the BLA is a key factor affecting the consolidation of conditioned fear memory, and this is mediated by changes in synaptic structural plasticity in the BLA. Further research found that the self-phosphorylation of calcium/calmodulin-dependent protein kinase II (CaMKII) at the Thr-262 site mediated by CCK2R leads to changes in post-synaptic density-95 (PSD-95) in amygdala neurons, causing alterations in synaptic structural plasticity in amygdala neurons; this study confirmed that CCK in the BLA influences the synaptic remodeling and structural plasticity of BLA neurons, promoting the consolidation of conditioned fear memory[27].
Research has confirmed that high concentrations of CCK and CCK mRNA exist in neurons within the BLA[38]. CCK-positive neurons in the amygdala have a strong influence on regulating the excitability of pyramidal neurons and synaptic integration; studies have also found that CCK neurons are involved in emotional regulation. Crosby et al.[49] found that CCK can alter the plasticity of GABA synapses in the dorsal nucleus of the hypothalamus, shifting from long-term depression (LTD) under physiological conditions to long-term potentiation (LTP), a process requiring activation of CCK2R; the latter acts on metabotropic glutamate receptors on astrocytes to trigger an increase in intracellular calcium concentration and release the gliotransmitter ATP, which subsequently acts on purinergic receptors on GABA to promote GABA release[4]. CCK8S is currently known as the strongest endogenous anti-opioid substance, and opioid peptides can enhance the effect of “presynaptic inhibition” mediated by GABA, while CCK8S can counteract this effect of opioid peptides. Research on CCK’s regulation of synaptic plasticity has been more focused on appetite and satiety signals, and further exploration of the relationship between CCK and synaptic plasticity in depression animal models is needed.
3.4 Circuit Mechanisms
Studies have found that bilateral injection of CCK8S into the ventral hippocampus reduces the percentage of open arm time (%OAT) and the percentage of open arm entries (%OAE) in rats, exhibiting anxiety-like behavior. Bilateral injection of the selective CCK2R antagonist LY225910 does not alter anxiety-related parameters, but using selective GABAAR agonists can dose-dependently increase %OAT and %OAE, indicating an anxiolytic effect, suggesting that GABAAR receptors are involved in the anxiety behavior induced by CCK8S in the ventral hippocampus[51].
Research shows that 5HT1B receptors on inhibitory neurons of the dentate gyrus in mammals can trigger treatment responses to antidepressants, which are activated presynaptically to inhibit GABA release, relieving inhibition on PV neurons and reducing the activity of granule cell neurons[52]. Whissell et al.[53] demonstrated that selective activation of CCK-GABA neurons can enhance memory and cognitive abilities. Using optogenetic methods, the specific role of CCK in the mPFC pathway was confirmed: CCK mediates anxiety symptoms in the mPFC-BLA projection, while CCK mediates depressive symptoms in the mPFC-NAc projection. The CMS rat model sensitizes cannabinoid receptor type 1 (CB1R) at the terminals of GABAergic neurons, leading to increased excitatory neurotransmission of CCK-GABA due to WIN (CB1 receptor agonist). The CSDS stress model selectively enhances the excitatory transmission of the CCK-D2 circuit in susceptible mice by reducing presynaptic CB1R; knocking out CB1R in the CCK-D2 circuit can increase synaptic activity and enhance stress sensitivity. Notably, selective inhibition of the CCK-D2 circuit or administering synthetic cannabinoids in the NAC can produce antidepressant-like effects; in CSDS-induced depression-susceptible mice, the excitatory synaptic transmission of the CCKBLA-D2NAc glutamate aversion circuit is significantly enhanced, and optogenetic inhibition of this circuit can effectively overcome depressive symptoms[38]. CCK coexists with dopamine in dopaminergic neurons in the mesolimbic system and nigrostriatal pathway, and CCK participates in regulating dopamine activity through CCK1R and CCK2R. CCK1R in the mid-ventral tegmental area can promote dopamine release, while CCK2R in the anterior part of the nucleus accumbens can inhibit dopamine release. Studies indicate that CCK’s influence on dopaminergic neurons through CCK1R and CCK2R may play an important role in the pathogenesis of mental disorders.[54].
4 Summary and Outlook
This article summarizes the relationship between CCK and depression, and organizes the role of CCK in depression from the aspects of HPA axis function, molecular mechanisms, synaptic mechanisms, and circuit mechanisms. In the narrative process from the relationship between CCK and depression to the mechanisms of CCK’s role in depression, the author identifies several existing issues: (1) There are differences in CCK levels in different brain regions in depression animal models; for example, stress increases CCK expression levels in the cortex of model rats, but CCK neuron expression decreases in the mPFC. Whether there is an interconnection between different brain regions remains to be studied. (2) In clinical studies, the correlation between plasma CCK levels in suicide attempters and depression rating scales is minimal, suggesting that there may be some directly related factors that mutually constrain the effects of CCK on patients with mental disorders, such as diet and certain psychological factors. (3) Currently, there is much research on the effects of CCK on synaptic plasticity in appetite, while research on its effects in depression needs to be deepened. This article provides an overall explanation of the connection between CCK and depression, offering a theoretical basis for further understanding the mechanisms of CCK’s role in depression.
