Click the “blue WeChat name” below the title to quickly follow
Source: Chinese Journal of Neurology, 2021, 54(7): 649-654
Authors: Chen Shuai, He Shuang, Pang Mi, Li Wei, Li Shujian, Zhang Jiewen
Abstract
Objective
This study reports the clinical manifestations and genetic characteristics of a family with familial encephalopathy with neuroserpin inclusions bodies (FENIB) to enhance understanding of this disease.
Methods
The proband presented with cognitive impairment and epilepsy at the Neurology Department of Henan Provincial People’s Hospital in June 2020. Detailed medical history, neurological examination, and family pedigree were conducted for the proband and family members. The proband underwent cranial magnetic resonance imaging (MRI), electroencephalogram (EEG), cerebrospinal fluid analysis, and whole exome sequencing. Sanger sequencing was performed for some family members for validation. Relevant literature was reviewed to analyze the characteristics of the disease.
Results
The proband was a 23-year-old female with cognitive impairment and epilepsy as the main manifestations, which gradually progressed. The proband’s mother and brother exhibited similar clinical symptoms in their youth and both passed away several years later. The cranial MRI of the proband showed extensive cortical atrophy bilaterally. Whole exome sequencing revealed a heterozygous missense mutation c.1013A>G (p.H338R) in the SERPINI1 gene, which encodes neuroserpin. This mutation has been previously reported as pathogenic. The clinical phenotypes within the family were consistent, and the gene mutation was co-segregated with the clinical phenotype.
Conclusion
In young patients with dementia combined with epilepsy and myoclonus, the possibility of FENIB caused by SERPINI1 gene mutation should be considered.
Neuroserpin (NSP) is a member of the highly conserved serine protease inhibitor (serpin) superfamily. Human NSP consists of 410 amino acids encoded by the SERPINI1 gene located at 3q26, and is widely distributed in the central nervous system, especially in regions such as the cerebral cortex, hippocampus, and amygdala. As a protease inhibitor, NSP is an important regulatory factor for protease activity in the central nervous system. In 1999, Davis et al. discovered abnormal eosinophilic inclusions within neurons in the pathological examination of two dementia families, identifying their components as abnormal deposits of NSP, and mutations in the SERPINI1 gene encoding NSP were found in the families. They first proposed the clinical pathological concept of familial encephalopathy with neuroserpin inclusions bodies (FENIB) and named these inclusions Collins bodies. Since then, several familial or sporadic cases have been reported in European and American populations. In 2018, Japanese scholars reported the first Asian FENIB family. To date, 12 cases of FENIB families or sporadic cases caused by six types of SERPINI1 gene point mutations have been reported worldwide. This article reports the first FENIB family in China, detailing its clinical, imaging, and genetic characteristics to enhance understanding of the disease.
Materials and Methods
1. Study Subjects
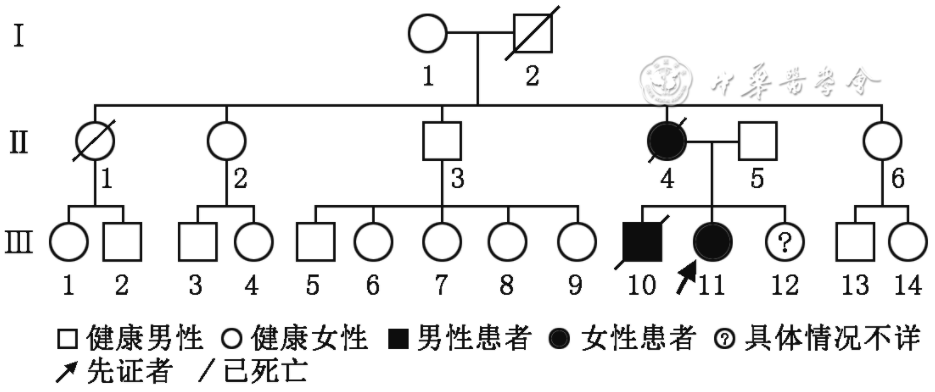
The proband was a 23-year-old female patient who visited Henan Provincial People’s Hospital (Zhengzhou University People’s Hospital) in June 2020. She was admitted due to “memory loss accompanied by epilepsy for 2 years, worsening in the last 3 days.” Family history revealed that the patient’s mother and brother had similar medical histories. We conducted detailed medical history inquiries, neurological examinations, and drew a family pedigree (Figure 1). Genetic testing was performed on the proband and some family members using peripheral blood.
Figure 1 Pedigree of familial encephalopathy with neuroserpin inclusions bodies. Ⅰ1 is 80 years old and healthy; Ⅰ2 died of cerebrovascular disease at the age of 72. Ⅱ1 died accidentally at the age of 40 without similar complaints. Ⅱ4 is the mother of the proband, who showed cognitive symptoms such as decreased ability in housework and memory at the age of 17, followed by frequent seizures, and died at the age of 30. Ⅱ3 has diabetes, cerebral hemorrhage, and infarction; Ⅱ2, Ⅱ5, Ⅱ6 are healthy. Ⅲ10 had frequent seizures since the age of 18, followed by cognitive impairment that gradually worsened, and died at the age of 26; Ⅲ11 is the proband; Ⅲ12 was fostered elsewhere since childhood and lost contact; Ⅲ1-9, Ⅲ13-14 are currently healthy.
Figure 1 Pedigree of familial encephalopathy with neuroserpin inclusions bodies. Ⅰ1 is 80 years old and healthy. Ⅰ2 died of cerebrovascular disease at the age of 72. Ⅱ1 died accidentally at the age of 40 without similar complaints. Ⅱ4 is the mother of the proband. At the age of 17, she presented with cognitive symptoms such as decreased housework ability and memory, mental-behavioral disturbance. Then she suffered frequent seizures and died at the age of 30. Ⅱ3 has diabetes, cerebral hemorrhage, and infarction. Ⅱ2, 5, 6 are healthy. Ⅲ10 had frequent seizures since the age of 18, followed by cognitive decline, and died at the age of 26. Ⅲ11 is the proband. Ⅲ12 had been fostered in another family and lost contact since childhood. Ⅲ1-9, 13-14 are currently healthy.
This study is a family study approved by the Ethics Committee of Henan Provincial People’s Hospital (Approval No: 2020 Ethics Review No. 76). Informed consent was obtained from the patient’s family for clinical data collection and genetic testing.
2. Research Methods
(1) Auxiliary Examinations
The proband underwent a comprehensive examination including blood routine, liver and kidney function, electrolytes, blood lipids, blood glucose, tumor markers, TORCH (Toxoplasma, Rubella virus, Cytomegalovirus, Herpes simplex virus type I/II IgM and IgG antibodies), vitamin B12 and folate levels, antinuclear antibodies and anti-soluble antigen antibodies, anti-neutrophil cytoplasmic antibodies, thyroid function tests, blood ammonia, lactate, infectious disease screening, autoimmune encephalitis antibody tests, cerebrospinal fluid analysis, and urine organic acid blood tandem mass spectrometry for metabolic disease screening. Imaging examinations included chest CT, cranial MRI plain scan with enhancement, video EEG, electromyography of limbs, and ultrasound of the thyroid, abdomen, and gynecology.
(2) Genetic Diagnosis
3 ml of peripheral blood was collected from the proband (Ⅲ11) and some relatives (Ⅰ1, Ⅱ2, Ⅱ3, Ⅱ5, Ⅱ6) for genomic DNA extraction. Considering the proband’s cognitive impairment and the possibility of mitochondrial disease, whole exome sequencing, copy number variations (CNV), and mitochondrial 16K testing were performed. The specific methods were as follows: First, the xGen® Exome Research Panel v1.0 capture probes from IDT were used for liquid hybridization with genomic DNA (gDNA) library sequences to enrich the target region DNA fragments, constructing a whole exome library covering the coding regions of 19,396 genes and some non-coding regions of the human genome, with a capture interval size of 51 Mb. High-throughput sequencing (PE150) was performed using the NovaSeq 6000 series sequencer from Illumina, with a target sequence coverage of no less than 99%. The raw data were quality controlled using fastp software, removing adapters and low-quality sequences. GenomeAnalysisTK (GATK v3.7) software was used to identify variant sites in the sample sequencing data compared to the reference genome, and ANNOVAR software was used for functional annotation of these variant sites. Mutations were filtered using public databases such as ExAC, 1000 Genomes Project, and ESP6500. For CNV, a control database was established using samples sequenced in the same batch to statistically normalize the coverage depth of each exon, analyze CNV, and verify its reliability. After identifying potentially pathogenic gene point mutations in the proband, Sanger sequencing was used for validation in the proband and some relatives.
Results
1. Clinical Data
The patient is a 23-year-old female farmer. She was admitted due to “memory loss accompanied by seizures for 2 years, worsening in the last 3 days.” Two years ago, her family noticed her slow responses, reduced speech, memory loss (forgetting things), blurred vision, and decreased daily living abilities. She also experienced episodes of altered consciousness, limb twitching, foaming at the mouth, and incontinence, with seizures lasting 10-30 minutes, and recovery of consciousness after about 2 hours. Similar symptoms recurred, often triggered by emotional agitation. Family members also noticed brief twitching of her lips and eyelids at night. She had previously visited a local hospital, where cranial CT suggested brain atrophy, and blood tests indicated microcytic hypochromic anemia, for which she was treated with antiepileptic drugs. Six months ago, the patient began to experience brief tremors throughout her body when using her hands or after contact, and her ability to hold objects became unstable, with further cognitive decline and purposeless pacing behavior, making daily living difficult. Three days ago, she experienced dizziness and was unable to walk, with more pronounced tremors in her limbs when exerting herself. She then visited our hospital, where she was admitted with a diagnosis of “epilepsy with cognitive impairment under investigation.” Past medical history: Hepatitis B virus carrier, otherwise unremarkable. Personal history: Full-term normal delivery, no birth injury or hypoxia, normal physical development since childhood, able to exercise, graduated from elementary school with poor grades, and poor ability in cooking and household chores. Family history: The patient’s mother and brother had similar manifestations; the mother fell ill at 17, presenting with dementia and epilepsy, and died at 30. The brother had onset of epilepsy at 18, followed by dementia, and died at 26.
Physical examination: No abnormalities in the internal medicine examination. The patient was conscious, right-handed, in a lethargic state, with poor speech comprehension, minimal spontaneous speech, and repetitive stereotyped movements. Orientation, calculation, memory, and judgment were poor. No abnormalities were found in cranial nerves. Limb muscle strength and tone were normal, bilateral lower limb tendon reflexes were active, and bilateral pathological signs were not elicited. Rough tests of deep and superficial sensations were normal, and coordination was difficult to cooperate with. Meningeal irritation signs were negative.
During hospitalization, the patient exhibited myoclonus in her limbs when exerting herself. She was treated with levetiracetam 0.5 g twice daily, coenzyme Q10, iron supplements, etc., with good control of seizures, and genetic testing was completed.
2. Auxiliary Examinations
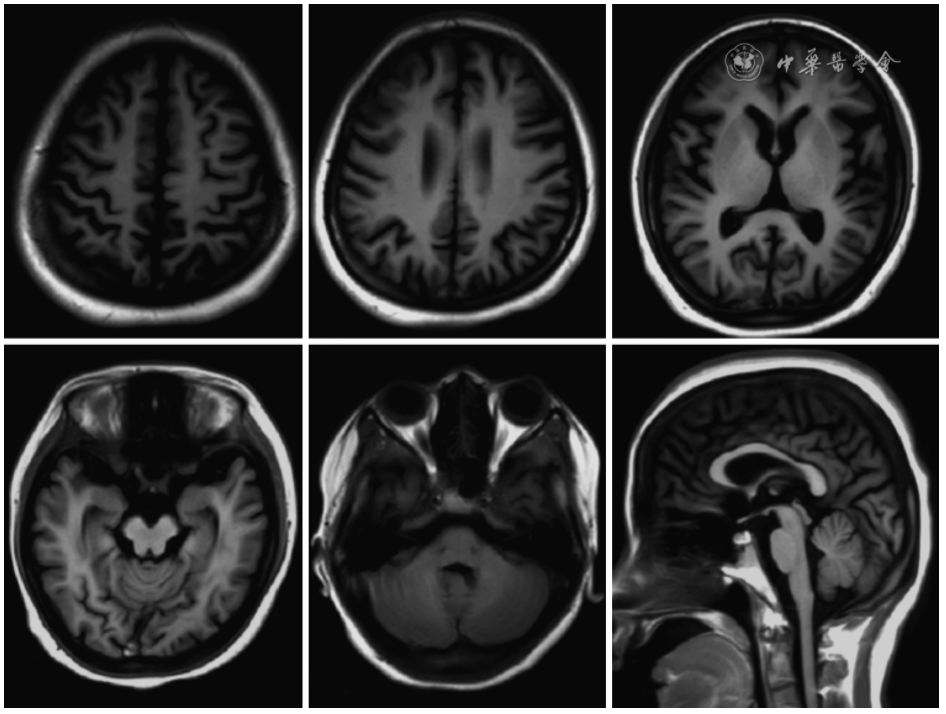
Blood routine showed white blood cells 4.48×109/L, red blood cells 4.66×1012/L, hemoglobin 85 g/L, mean corpuscular volume 68.9 fl, and platelets 126×109/L. Hepatitis B qualitative: HBsAg (+), HBeAb (+), HBcAb (+). No significant abnormalities were found in other tests. Genetic metabolic disease mass spectrometry screening was negative. Thyroid ultrasound showed bilateral follicular hyperplasia, and abdominal and pelvic ultrasound showed a cystic echo in the right ovary. Chest CT showed no abnormalities. Cranial MRI plain scan + enhancement: mild brain atrophy (Figure 2). Video EEG (the patient was already on levetiracetam and alprazolam during the examination, monitored for 10 hours without clinical seizures): In the awake eyes-closed state, the background rhythm in the occipital region of both hemispheres was predominantly theta waves at 5-7 Hz, with slightly more low-amplitude beta waves, and visual response inhibition was poor. Over-breathing did not significantly increase slow waves in all leads, and occasional single spikes and slow waves were observed. Electromyography of the limbs and nerve conduction showed no abnormalities.
Figure 2 T1WI sequence of brain magnetic resonance imaging of the patient with familial encephalopathy with neuroserpin inclusions bodies, which showed whole cortex atrophy and thin corpus callosum at the posterior part.
Figure 2 T1WI sequence of brain magnetic resonance imaging of the patient with familial encephalopathy with neuroserpin inclusions bodies, which showed whole cortex atrophy and thin corpus callosum at the posterior part.
3. Genetic Characteristics
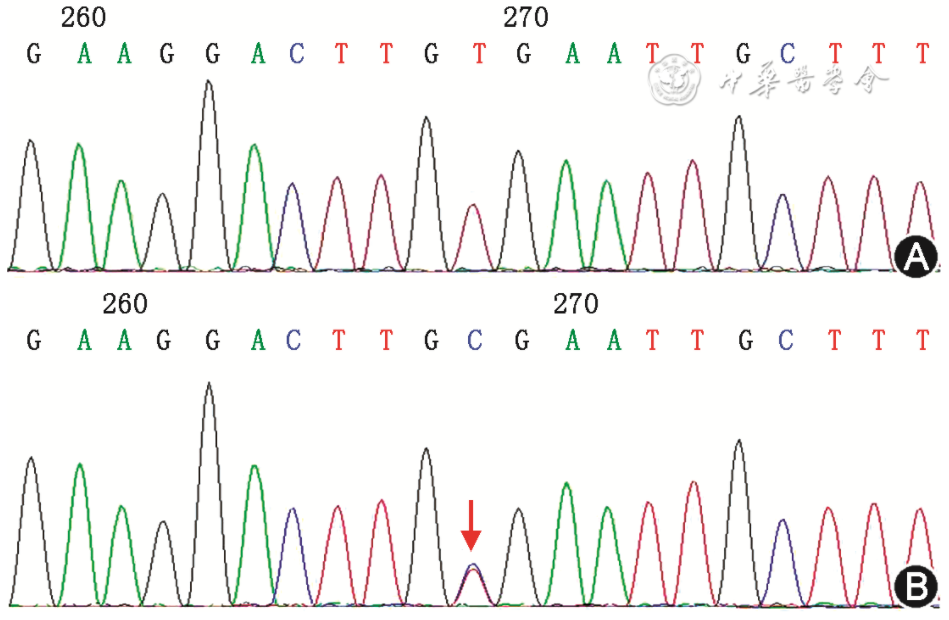
Whole exome sequencing was performed on the proband, with an average sequencing depth of 120X and a coverage rate of 99.45% above 20X. A heterozygous mutation c.1013A>G was found in exon 7 of the SERPINI1 gene (Figure 3), causing a change in the amino acid sequence to p.H338R (NM_005025). No pathogenic CNVs related to the clinical phenotype of the proband were found. The mitochondrial 16K test results did not reveal any hotspot mutations related to the clinical phenotype of the proband.
Figure 3 Sanger sequencing of SERPINI1 gene in the proband with familial encephalopathy with neuroserpin inclusions bodies and her family members. A: the uncle of the proband (Ⅱ3) without mutation; B: the proband, the arrow showing the 1013T>C heterozygous mutation (reverse A>G mutation).
The H338R mutation site is absent in the normal control populations of the gnomAD database, ESP6500 Exome Project database, 1000 Genomes Project, and ExAC database. Sanger sequencing further confirmed the p.H338R mutation in the proband, and this mutation was not found in family members Ⅰ1, Ⅱ2, Ⅱ3, Ⅱ5, Ⅱ6. Functional prediction software results: SIFT (0.026, harmful); PolyPhen-2 (0.482, possibly harmful); MutationTaster (1.0, pathogenic); GERP++ (5.24, conserved). Literature review indicates that this mutation has been previously reported abroad and confirmed by brain tissue pathology as a pathogenic mutation for FENIB. Patients in this family exhibited early-onset, gradually worsening dementia-epilepsy syndrome, with clinical phenotypes consistent with FENIB. Family pedigree analysis indicated that the inheritance pattern is consistent with autosomal dominant inheritance of FENIB, and the gene mutation co-segregated with the clinical phenotype.
Discussion
The main clinical features of this patient include early-onset, gradually worsening cognitive impairment and epilepsy syndrome, with a positive family history. The epilepsy primarily manifested as generalized tonic-clonic seizures, accompanied by myoclonus (or myoclonic epilepsy), with dementia and epilepsy appearing almost simultaneously. Cranial MRI showed mild brain atrophy, and other routine examinations showed no specific findings. Family analysis suggested that the disease may be inherited in an autosomal dominant or maternal manner. Differential diagnosis should consider epileptic encephalopathy caused by frequent seizures and encephalopathy presenting with both cognitive and epileptic symptoms, such as genetic metabolic and neurodevelopmental disorders. Since the patient presented with simultaneous dementia and epilepsy, with infrequent seizures, it does not align with epileptic encephalopathy. Therefore, hereditary metabolic diseases should be qualitatively considered, especially neurodegenerative diseases characterized by epilepsy, progressive cognitive impairment, and myoclonus, such as progressive myoclonic epilepsy. Progressive myoclonic epilepsy diseases inherited in an autosomal dominant or maternal manner include KCNC1 gene-related myoclonic epilepsy, myoclonic epilepsy with ragged red fibers (MERRF), dentatorubral-pallidoluysian atrophy (DRPLA), and FENIB. Therefore, genetic testing was selected for whole exome, CNV, and mitochondrial gene testing, which ultimately identified the FENIB-related gene mutation, confirming the diagnosis.
FENIB is caused by mutations in the SERPINI1 gene, leading to conformational changes in NSP, which spontaneously form polymers that ultimately aggregate into inclusions within brain neurons, causing pathology. In 1999, Davis et al. reported the first family with clinical features of cognitive impairment starting around the age of 50, carrying the SERPINI1 gene S49P point mutation, with pathological findings showing inclusion deposits in the cortex and deep nuclei (such as the substantia nigra). The second family had dementia and epilepsy onset in their 20s to 30s, carrying the S52R point mutation, with pathological findings also showing inclusions within neurons. In 2000, Takao et al. discovered another S52R mutation family, with clinical phenotypes of myoclonic epilepsy starting around the age of 25. Detailed pathological examinations of patients in this family found Collins bodies only deposited in brain and spinal cord tissues, with no Collins bodies found in skin, liver, lung, spleen, pancreas, muscle, pituitary, adrenal glands, or thyroid.
Collins bodies are located in the rough endoplasmic reticulum, measuring approximately 5-50 μm in diameter, often located perinuclear or dendritic, and due to their large size, the nucleus can be pushed to the periphery. Collins bodies are positive for periodic acid-Schiff (PAS) staining and are resistant to amylase. Collins bodies are similar in size to Lafora bodies found in progressive myoclonic epilepsy, both being PAS positive, but they have distinct differences under electron microscopy and can be differentiated by immunohistochemistry. In terms of distribution, Lafora bodies can be widely found in the retina, skeletal and cardiac muscle, liver, skin, and cerebellum, while Collins bodies are only found in the central nervous system, especially in the cortex, and are rarely found in the cerebellum.
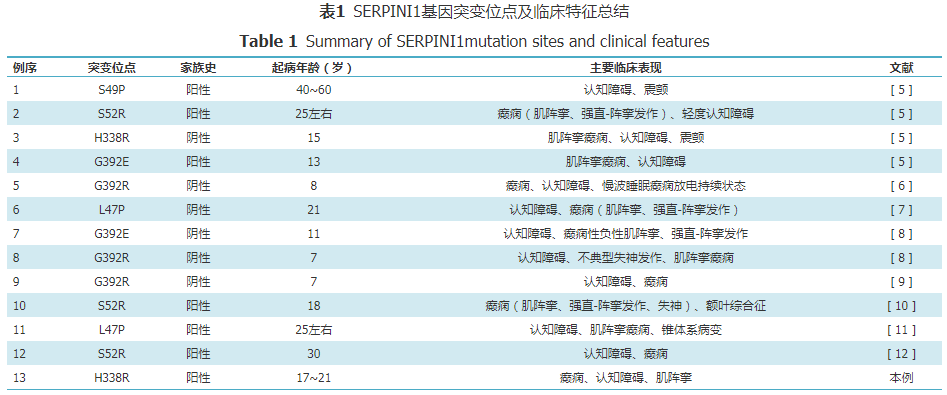
Currently, 12 cases of FENIB families or sporadic patients have been reported worldwide, all carrying SERPINI1 gene mutations, mainly from European and American populations. Early reported cases were mostly familial, but sporadic cases have gradually been discovered. Therefore, naming it hereditary familial encephalopathy with neuroserpin inclusions bodies may be more accurate. In 2018, Japan reported the first FENIB family in Asia. A review of the relevant literature shows that six types of SERPINI1 gene mutation sites have been found in FENIB patients, namely L47P, S49P, S52R, H338R, G392E, and G392R. Different point mutations exhibit relatively specific clinical and pathological features. Patients with the S52R mutation often present with myoclonus and epilepsy in their teens to 30s, followed by cognitive deterioration. Patients with the S49P mutation often present with progressive cognitive impairment in their 40s. Patients with H338R, G392E, or G392R mutations have an earlier onset age, at 15, 13, 11, and 7-8 years, with clinical manifestations including epilepsy, dementia, and prolonged slow-wave sleep epileptic discharges, with particularly severe conditions. A patient with the L47P mutation from Belarus presented with progressive myoclonic epilepsy with onset in adolescence, followed by cognitive impairment, without a positive family history. The L47P mutation family reported in Japan had the first symptoms of cognitive impairment compared to peers since childhood, followed by the onset of epilepsy. Davis et al. found that the pathogenicity strength of different mutations based on the amount of Collins body deposits was as follows: G392E>H338R>S52R>S49P. This pathological result is consistent with clinical and protein biological function predictions, suggesting that patients with G392E and H338R mutations have earlier onset and more severe conditions. Takehara et al. studied the polymerization pathways of wild-type neuroserpin and mutant S49P and H338R and found that the S49P mutant can fold into a conformation similar to the wild type but at a slower rate, while the H338R mutant is more difficult to fold into the natural conformation and forms more polymers.
This case is the second patient with the H338R mutation, with clinical manifestations similar to the first patient with the H338R mutation, both presenting with early-onset dementia and epilepsy. The first patient with the H338R mutation had no clear positive family history. The family history and negative gene testing results of the first and second generations in this family suggest that the mother may have a de novo mutation. A limitation of this case is that during video EEG monitoring, the patient did not exhibit myoclonus, making it unclear whether her myoclonus was epileptic or non-epileptic cortical myoclonus. Due to widespread cortical involvement, both could theoretically occur.
The molecular conformation of NSP is variable, stably binding to its target molecules, and this conformational change is easily affected by point mutations. According to protein structure models, the region most susceptible to mutation is located between the 3rd and 5th strands of the β-sheet A, which is a key area for NSP conformational transition. The conformational ability of mutated NSP is facilitated, leading to ordered extension and polymer formation. One of the homologous proteins of NSP is α1-antitrypsin, whose conformational changes can lead to the formation of inclusions in hepatocytes and cirrhosis in adolescents. The S49P and H338R mutation sites of NSP correspond to the S53F and H334D mutations of α1-antitrypsin, respectively. The exact pathogenic mechanism of NSP abnormal deposition is still unclear. It may be due to the toxic gain of function caused by the deposition of inclusions within neurons or related to the loss of normal physiological function of NSP. Under physiological conditions, NSP is an inhibitor of tissue-type plasminogen activator (tPA), which plays an important role in neuronal remodeling and maintenance, and is related to long-term potentiation and seizure activity. Animal model studies have found that NSP may exert an anti-epileptic effect by inhibiting tPA.
Currently, treatment for FENIB is mainly symptomatic, with no radical cure available, and the survival period after onset is 3-10 years. This case suggests that clinicians should pay attention to family history inquiries and the possibility of FENIB in young patients with early-onset dementia combined with epilepsy and myoclonus. Due to the difficulty of obtaining brain tissue pathology samples and the lack of Collins bodies in peripheral tissues, genetic testing is particularly important.
References omitted



