If you like it, please follow me for more updates.
Carbon dots (CDs) are promising luminescent nanomaterials with advantages such as small size, good biocompatibility, photostability, and low toxicity. They have great potential in the fields of bioimaging and biosensing. Currently, various CDs with efficient blue and green fluorescence emission have been widely reported, while the success rate of preparing red-emitting CDs is relatively limited. It is well known that longer wavelength light can penetrate deeper biological tissues, avoiding interference from the organism’s own fluorescence, thereby increasing contrast and spatial resolution. Deep red (wavelengths greater than 650 nm) and near-infrared (NIR, 780-2500 nm) emitting CDs are of great significance in deep tissue bioimaging.
It is known that a large number of surface functional groups significantly affect the fluorescence properties of CDs. On one hand, these functional groups enable CDs to easily bind with various matrices to form CDs-based composites, enhancing the fluorescence of CDs in the solid state. On the other hand, these surface groups can interact with the surrounding environment (such as solvents) to exhibit regulated emission and solvent chromogenic properties. This provides an opportunity to explore CDs-based composites that emit deep red/near-infrared light in both solid and aqueous phases.
Overview of the Full Text
Based on this, Professors Li Jiyang and Yu Jihong from Jilin University first prepared CDs with interesting solvent-dependent and two-photon fluorescence emission using a simple solvothermal method. Detailed characterization revealed that the carboxyl functional groups on the CDs interact with electron-donating groups in the solvent through n→π* interactions, leading to an increase in the energy density of the CDs, a decrease in energy levels, and a redshift in the luminescence as the electron-donating ability of the solvent increases. Inspired by this discovery, deep red fluorescence emission was achieved in solid and aqueous phases by confining CDs using amino-modified mesoporous silica nanoparticles (MSNs) with suitable pore sizes and low biotoxicity through host-guest n→π* interactions. The prepared CDs@EDA-MSN composites exhibited efficient fluorescence at a wavelength of 650 nm, low toxicity, and good biocompatibility, showing great potential in bioimaging applications.
This work was published in Nano Research under the title “Confining Carbon Dots in Amino-functionalized Mesoporous Silica: n→π* Interaction Triggered Deep-red Solid-state Fluorescence”.
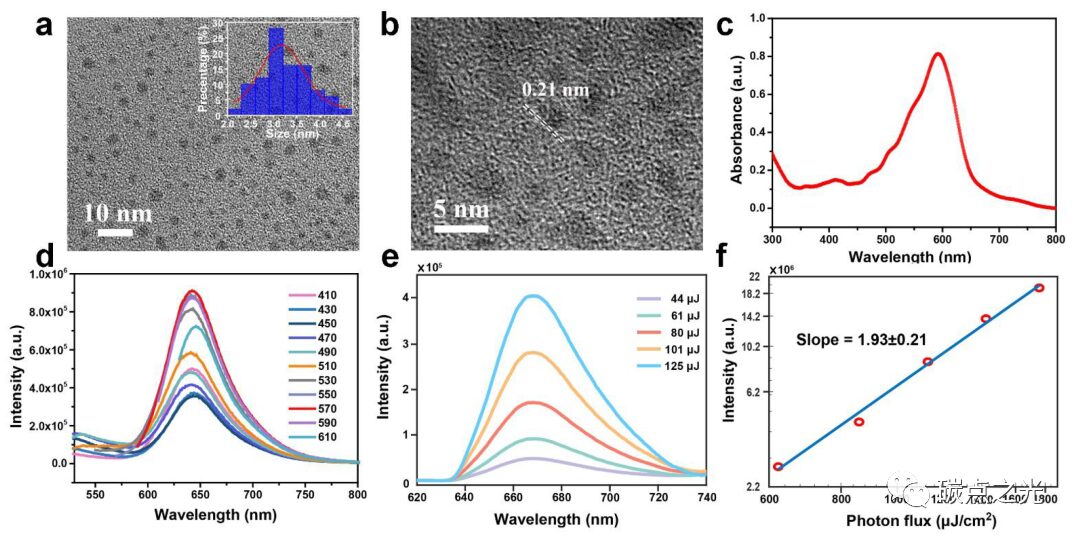
Figure 1. Morphology and optical properties of CDs. (a) TEM and (b) HRTEM images; (c) UV-visible absorption spectrum; (d) fluorescence spectrum under 410 – 610 nm excitation; (e) two-photon fluorescence spectrum under different powers of 800 nm femtosecond laser; (f) relationship between the two-photon fluorescence intensity of CDs and the laser photon flux.
Using citric acid (CA), urea, and DMF as raw materials, CDs were prepared by a simple solvothermal method, and purified using silica gel column chromatography. Transmission electron microscopy (TEM) images showed the quasi-spherical morphology and narrow size distribution (about 3.2 nm) of the synthesized CDs (Figure 1a). High-resolution TEM images revealed distinct lattice fringes associated with the graphite (100) diffraction plane (Figure 1b). When CDs are dispersed in DMF solvent, they exhibit a very broad absorption band, nearly covering the entire visible spectrum, with maximum absorption concentrated at 580 nm (Figure 1c). Under UV excitation, the CDs solution emits strong red fluorescence, with an emission peak around 640 nm, and the emission peak remains essentially unchanged as the excitation peak varies from 410 nm to 610 nm, exhibiting excitation-independent behavior (Figure 1d). Additionally, under the excitation of an 800 nm laser, the CDs solution exhibits deep red fluorescence emission centered at 670 nm (Figure 1e). The emission peak remains unchanged, but the fluorescence intensity shows a quadratic relationship with the laser photon flux, confirming the two-photon fluorescence capability of the CDs (Figure 1f).
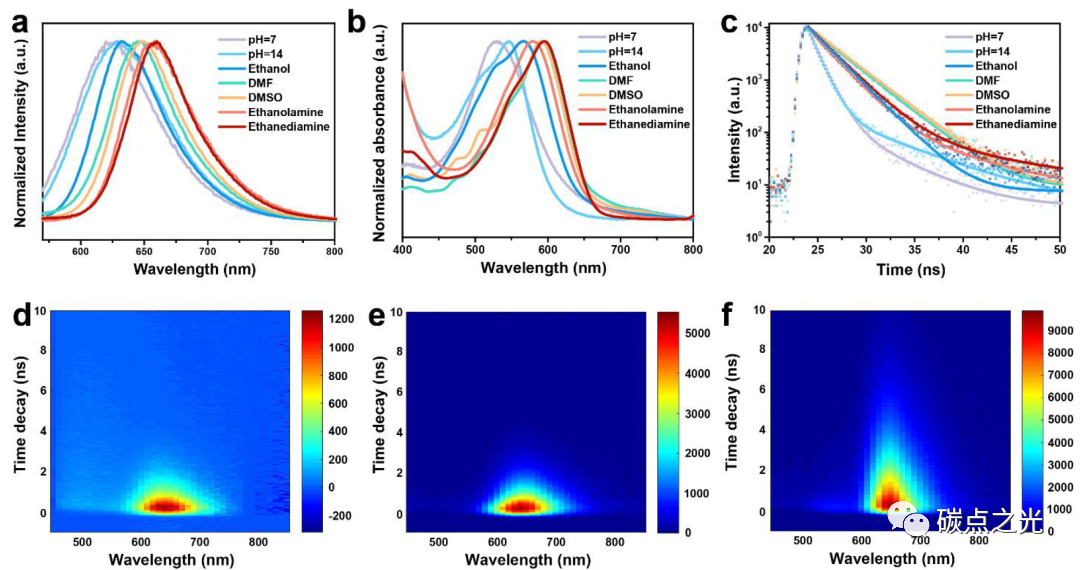
Figure 2. Solvent-related PL characteristics of CDs. (a) Fluorescence spectra of CDs in different solvents; (b) UV-vis spectra; (c) PL decay curves; (d-f) time-resolved decay spectra of CDs in water, NaOH, and DMF.
It is well known that CDs with deep red emission in water are particularly significant for deep tissue bioimaging. Considering the solvent chromogenic properties of CDs, where the absorption and emission spectra show a strong dependence on the type of solvent, the solvent-responsive fluorescence behavior of the prepared CDs was evaluated. As shown in Figure 2a, significant fluorescence changes were observed in different solvents. When dispersed in H2O solution, the CDs exhibited red emission centered at 625 nm, with a quantum yield (QY) of 2.57% (pH=7). When NaOH was added to water at pH=14, this emission slightly shifted to 628 nm, but the QY increased to 18.55%. However, with further increases in the concentration of NaOH in water, the emission peak of CDs no longer redshifted. Due to the poor solubility of CDs in acidic environments, the fluorescence emission of CDs became too weak for capture by fluorescence spectrophotometers. The UV-visible absorption spectrum (Figure 2b) shows a significant redshift of the absorption peak of CDs in organic solvents containing electron-donating groups compared to water and NaOH solutions, consistent with the above PL analysis. From the PL decay curves of CDs in different solvents shown in Figure 2c, it can be seen that the fluorescence lifetime of CDs in organic solvents is prolonged. Time-resolved fluorescence spectra of CDs in different solvents further confirm this (Figure 2d-f).
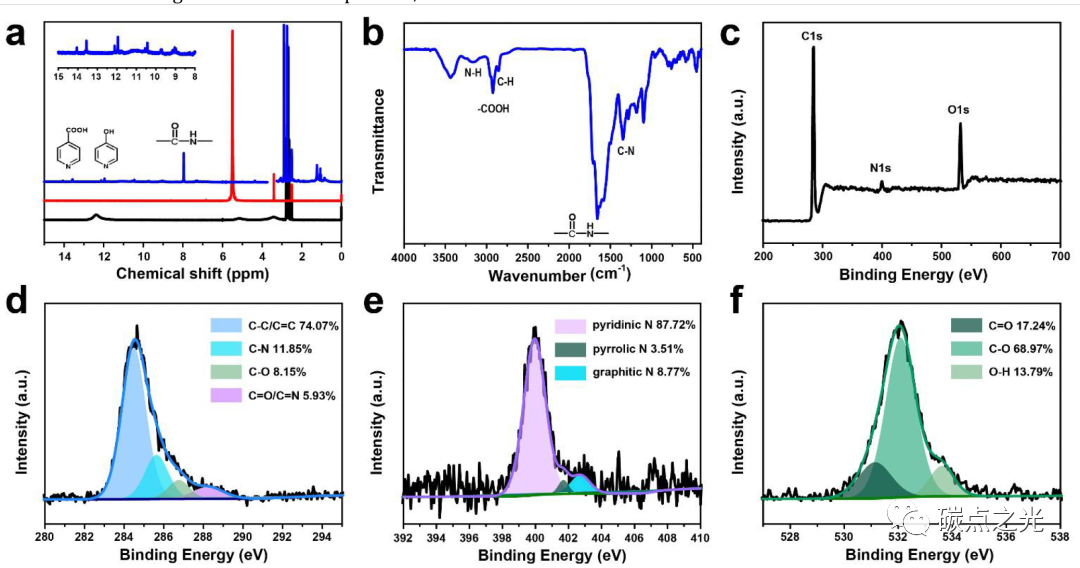
Figure 3. Structural characteristics of CDs. (a) 1H NMR spectra of CDs (blue line), urea (red line), and citric acid (black line); (b) FTIR spectrum of CDs; (c) XPS spectrum of CDs; (d-e) high-resolution C 1s, N 1s, and O 1s XPS fitting results and relative contents of each component.
The structure and chemical composition of CDs were studied using 1H NMR, FTIR, and XPS, revealing the mechanism of the solvent-dependent PL behavior of CDs. To compare the changes in functional groups during the solvothermal process of CDs, 1H NMR analysis was also performed on the raw materials (citric acid/urea) dispersed in DMSO-d6. As shown in Figure 3a, the chemical shifts of the amino group in urea (5.5 ppm) and -COOH in citric acid (12.4 ppm) shift to lower field in CDs (7.9 ppm and 13.6 ppm). It is known that when bonding with electron-affine π-conjugated groups (such as benzene or pyridine), the chemical shifts of functional groups move to lower fields due to the deshielding effect. Therefore, the authors speculate that the cross-linking polymerization of the raw materials during the formation of CDs results in the formation of a large π-conjugated structure connected with amino and carboxyl groups. Additionally, the 1H chemical shifts located at 9-12 ppm are attributed to -OH groups connected to the π-conjugated structure. Residual water, solvent DMSO-d6, and C-H bonds show 1H signals at 3.3, 2.5 ppm, and 0-3 ppm, respectively. In the FTIR spectrum (Figure 3b), typical aromatic C=C stretching vibration peaks and aromatic C-H bending vibration peaks are found at 1579 and 1000-1300 cm-1, respectively. Moreover, N-H and C=O stretching vibration peaks representing amide bonds are present at 3164 cm-1 and 1655 cm-1. The C-N stretching vibration peak of aromatic amines is located at 1349 cm-1, indicating that during the polymerization process, the -NH2 groups in urea are converted into amide bonds and aromatic amines in CDs. Additionally, O-H stretching vibrations of water (3435 cm-1), carboxyl groups (2923 cm-1), and alkyl chains (2847 cm-1) are also observed. The XPS results of CDs (Figure 3c) are highly consistent with the above structural features. In the full spectrum, the atomic ratios of C, N, and O elements are observed to be 63.39%, 6.56%, and 30.05%, respectively. In the high-resolution XPS spectrum, the C 1s peak is deconvoluted into four contributions (Figure 3d): C-C/C=C bonds (284.5 eV), C-N bonds (285.6 eV), C-O bonds (286.7 eV), and C=N/C=N bonds (288.2 eV); the N 1s spectrum indicates that pyridine nitrogen (400.0 eV) is dominant (Figure 3e); the O 1s peak corresponds to C=O bonds (531.2 eV), C-O bonds (532.1 eV), and O-H bonds (533.6 eV) (Figure 3f), indicating the presence of -COOH and -OH functional groups on CDs. From the above analysis, it can be concluded that during the solvothermal process, the amino groups in urea react with the -COOH in citric acid to form amide bonds, which further polymerize to generate a pyridine N-doped π-conjugated carbon core; the remaining -COOH and -OH proton groups in citric acid combine with the π-conjugated carbon core to form surface functional groups that play an important role in interactions with electron-donating solvents.
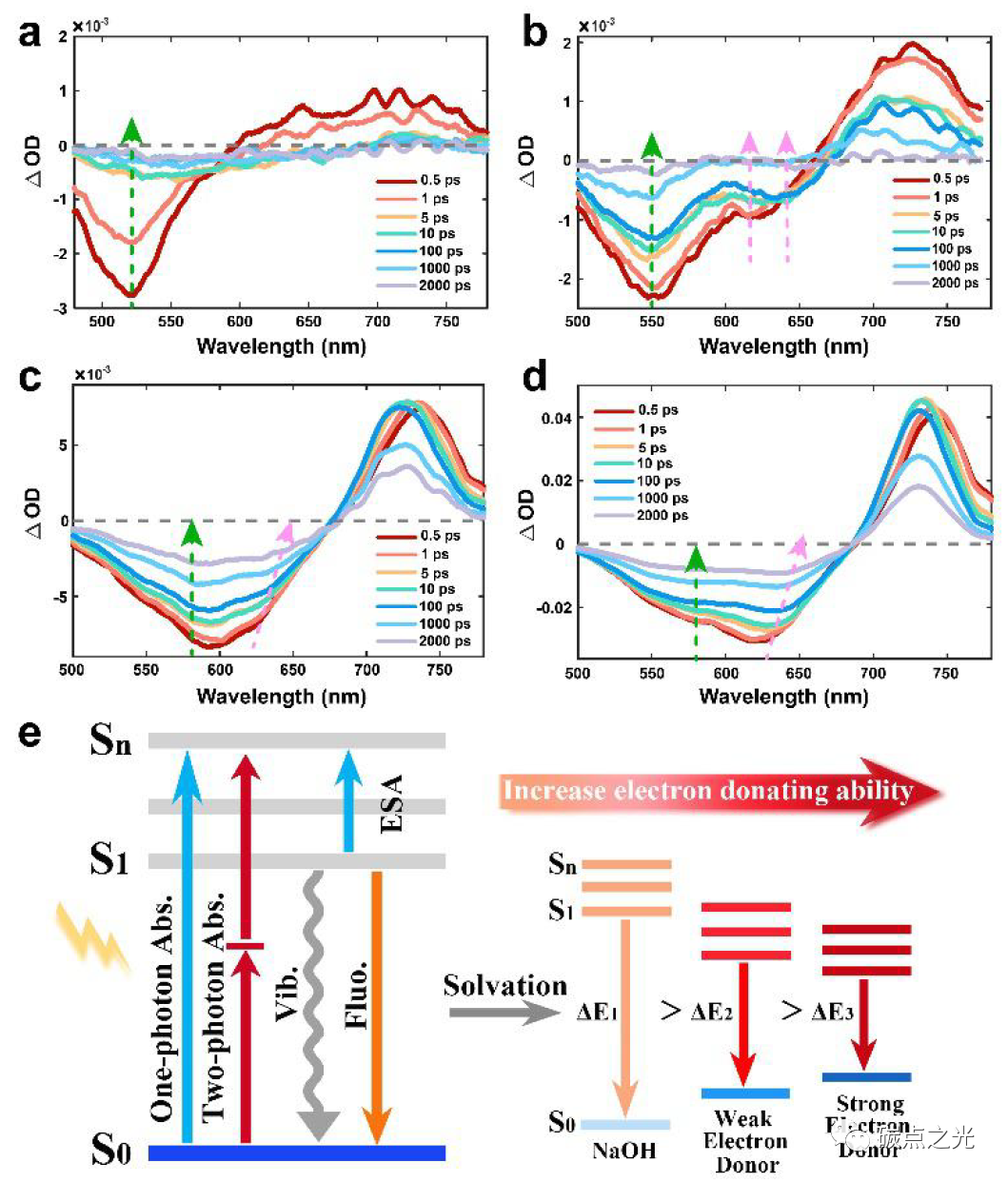
Figure 4. PL mechanism of CDs in different solvents. TA spectra of CDs dispersed in H2O, NaOH, DMF, and ethylenediamine showing changes at delay times from 0.5 ps to 2 ns; (e) energy levels and fluorescence decay schematic of CDs in solvents with different electron-donating abilities.
To gain deeper insights into the structure-induced variable luminescence process of CDs in different solvents, the transient absorption (TA) spectra of CDs in H2O, NaOH, and organic solvents were studied. As shown in Figure 4, negative signals represent ground state bleaching (GSB) and stimulated emission (SE), while positive signals correspond to excited state absorption (ESA). In H2O (Figure 4a), under excitation at 400 nm, CDs exhibit a negative peak from 450 to 600 nm, corresponding to the GSB peak according to the UV-vis absorption spectrum, while the SE peak is not obvious in water due to weak PL emission. In NaOH solution (Figure 4b), two negative peaks centered at 550 nm and 620 nm are observed, corresponding to GSB and SE signals of CDs, respectively. Notably, the SE peak at 620 nm rapidly shifts to 645 nm after 1 ps, which may be due to the higher electron density of -COOH groups on CDs in NaOH, leading to a reduction in the energy bandgap and a resultant redshift in emission. In DMF solvent (Figure 4c), as the detection time increases, the negative absorption peak at 600 nm gradually splits into two negative absorption peaks at 565 nm and 625 nm, corresponding to GSB and SE peaks, respectively. In ethylenediamine solvent (Figure 4d), two negative peaks for GSB and SE appear at 575 nm and 630 nm. Unlike the PL behavior in H2O and NaOH solutions, the SE peaks in DMF and ethylenediamine gradually split and redshift from 1 ps to 2000 ps. Additionally, a positive peak of ESA is observed at 700-800 nm in all solvents, indicating that the excited carriers absorb the detection light and migrate to higher energy levels. In summary, we conclude that in organic non-protonic alkaline solvents (such as DMF, DMSO, ethanolamine, and ethylenediamine), electron-donating groups such as amino groups interact with the -COOH groups on CDs, where the unique lone pair electrons in the nitrogen atom transfer to the CDs through donor-acceptor n→π* interactions, resulting in an increase in the energy density of CDs and a redshift in PL emission.
Based on the information gathered from the above analysis, the luminescence process of CDs in different solvents can be summarized as follows (Figure 4e): first, CDs are excited by the excitation light, rapidly forming thermally activated carriers within tens of femtoseconds; this thermally activated carrier relaxes from the Sn state to the S1 excited state (1-3 ps) through an internal conversion process. Subsequently, some excitons release energy through non-radiative thermal processes (Vib). In H2O solution, the surface functional groups of CDs exhibit a weak hydrolysis process, and carriers decay directly from S1 to S0 through radiative recombination, emitting fluorescence. In NaOH solution, the -COOH groups on CDs dissociate to COO–, leading to an increase in energy density, a decrease in energy levels, and a resultant redshift in fluorescence. In organic solvents, CDs are surrounded by electron-donating functional groups such as amino groups, where the lone pair electrons in the nitrogen atom can transfer to the carbonyl groups on CDs through n→π* interactions, thereby increasing the energy density of CDs, reducing the energy gap between S1 and S0, and promoting redshifted fluorescence emission. As the electron-donating ability of the solvent increases, the PL emission of CDs gradually redshifts. Consequently, CDs in ethylenediamine solvent exhibit strong deep red fluorescence emission at 660 nm with a QY of 43.41%, while CDs in DMSO show deep red emission near 648 nm with a QY as high as 63.81%.
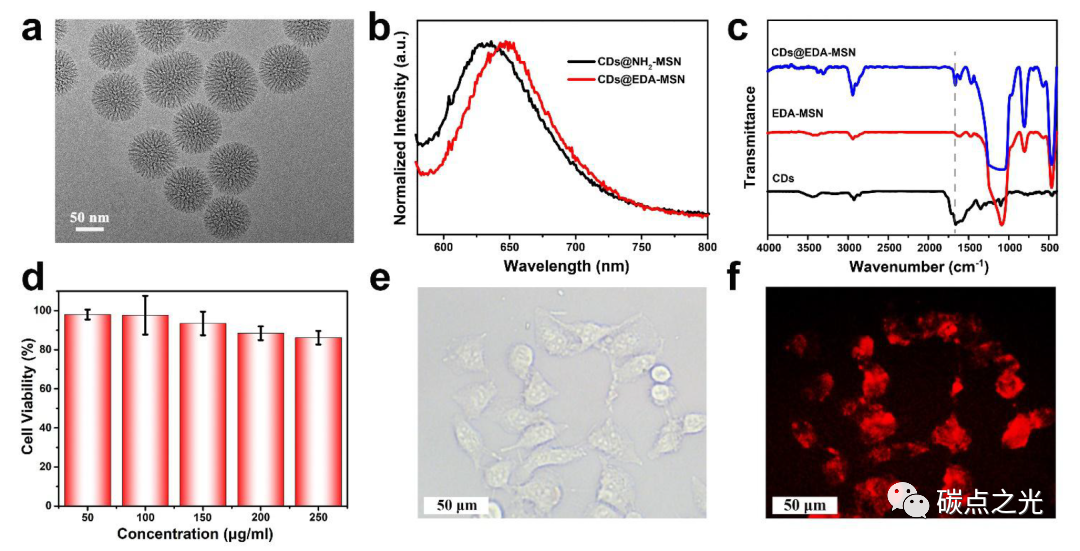
Figure 5. Confined CDs in amino-modified mesoporous silica and their applications in bioimaging. (a) TEM image of mesoporous silica; (b) emission spectra of CDs@NH2-MSN and CDs@EDA-MSN under 550 nm excitation; (c) FTIR spectra of CDs, EDA-MSN, and CDs@EDA-MSN; (d) cell viability of HeLa cells in the presence of different concentrations of CDs@EDA-MSN complexes; bioimaging of HeLa cells under green light stimulation with 250 μg/mL CDs@EDA-MSN (e) and bright field (f).
As shown in Figure 5a, the prepared matrix is dendritic mesoporous silica with an average pore size of 6 nm and a particle size of about 90 nm. Two amino groups with different electron-donating abilities, 3-aminopropyltriethoxysilane and 3-(2-aminoethylamino)propyltriethoxysilane, were used to modify the matrix, and then CDs were loaded through hydrogen bonding interactions, resulting in the composites named CDs@NH2-MSN and CDs@EDA-MSN, respectively. Taking the CDs@EDA-MSN composite as an example, the FTIR spectrum (Figure 5c) shows the presence of amide bonds of CDs in the CDs@EDA-MSN composite (dashed line), indicating the successful loading of CDs into the silica. As expected, the prepared CDs-based composites exhibit strong red fluorescence in the solid state. From Figure 5b, it can be seen that under 550 nm visible light excitation, the CDs@NH2-MSN solid powder emits red fluorescence at 633 nm, while the CDs@EDA-MSN powder emits deeper red fluorescence at 650 nm. This result confirms that the emission of CDs redshifts with the increase in the electron-donating ability of the surrounding environment. Additionally, mesoporous silica as a host matrix can enhance the stability of CDs, preventing fluorescence quenching caused by aggregation of CDs in the solid state. This matrix also facilitates deep red fluorescence emission of CDs in water, which is beneficial for applications in bioimaging and lighting. To further explore applications, CDs@EDA-MSN composite nanoparticles were uniformly dispersed in water to study their biotoxicity (Figure 5d). The cytotoxicity of HeLa cells was evaluated using the MTT assay. After 24 h of incubation with low concentrations of CDs@EDA-MSN solution (50 ~ 150 μg/mL), the cell survival rate reached over 90%. Even when 250 μg/mL of the complex was added to the HeLa cell culture medium, the cell survival rate remained above 80%, demonstrating that high levels of CDs@EDA-MSN have extremely low biotoxicity. Therefore, CDs@EDA-MSN composites are suitable for use as cellular imaging dyes. Bioimaging results (Figure 5e, f) show that the composite nanoparticles do not significantly affect cell morphology and emit bright red fluorescence under green light illumination, effectively shielding the blue fluorescence emitted by the organism itself. Notably, compared to the CDs reported in the literature that exhibit deep red fluorescence in organic solvents but weak fluorescence signals in water, the CDs@EDA-MSN composites exhibit bright deep red fluorescence in both solid and liquid states, greatly promoting their applications in bioimaging and lighting. It should be noted that in vivo bioimaging requires more refined design to further enhance the brightness and targeting ability of the composites, thereby achieving broader application prospects. Moreover, monodisperse silica microspheres, due to their rich hydroxyl groups on the skeleton, are easily modified by certain functional groups (such as amino, carboxyl, and thiol). Therefore, this MSN encapsulation method has broad applicability for developing universal CDs@MSN composites.
This work prepared a red-emitting CDs and systematically studied the interactions between CDs and solvents. Time-resolved spectroscopy and femtosecond TA techniques indicate that when CDs are surrounded by electron-donating groups in organic solvents, the fluorescence emission of CDs undergoes a redshift, where the lone pair electrons of the electron-donating groups in the solvent transfer to the carboxyl groups on CDs through n→π* interactions, thereby increasing the overall energy density of CDs and lowering energy levels. The results show that CDs exhibit deep red (660 nm) fluorescence emission in ethylenediamine with a QY of 43.41%, and high red emission in DMSO with a QY as high as 63.81%. Furthermore, by confining CDs within pre-designed amine-modified mesoporous silica, the host-guest structure strategy of n→π* interactions effectively promotes the occurrence of deep red-emitting CDs in both solid and aqueous phases. The prepared composites exhibit good biocompatibility, strong penetration, and bright deep red fluorescence for bioimaging. This work not only provides an in-depth understanding of the specific mechanisms of CDs emission processes in solvents but also offers new prospects for achieving deep red fluorescence emission based on CDs in both liquid and solid states, which can be used for future biomedical applications and optoelectronic devices.
Reference link: https://doi.org/10.1007/s12274-022-4829-x
We welcome submissions to the Carbon Dots Light WeChat public account. Submission email:
Click “Read the original text” to directly access the article above.